Kostniakomięsak
| ||
 Obraz histologiczny kostniakomięsaka | ||
| ICD-10 | C40 Nowotwór złośliwy kości i chrząstki stawowej kończyn | |
| C40.0 | Łopatka i kości długie kończyny górnej | |
| C40.1 | Kości krótkie kończyny górnej | |
| C40.2 | Kości długie kończyny dolnej | |
| C40.3 | Kości krótkie kończyny dolnej | |
| C40.8 | Zmiana przekraczająca granice jednego umiejscowienia w obrębie kości i chrząstki stawowej kończyn | |
| C40.9 | Kości i chrząstki stawowe kończyn, umiejscowienie nieokreślone | |
| ICD-10 | C41 Nowotwór złośliwy kości i chrząstki stawowej o innym i nieokreślonym umiejscowieniu | |
| C41.0 | Kości czaszki i twarzy | |
| C41.1 | Żuchwa | |
| C41.2 | Kości kręgosłupa | |
| C41.3 | Żebra, mostek i obojczyk | |
| C41.4 | Kości miednicy, kość krzyżowa i guziczna | |
| C41.8 | Zmiana przekraczająca granice jednego umiejscowienia w obrębie kości i chrząstek stawowych | |
| C41.9 | Kości i chrząstki stawowe, umiejscowienie nieokreślone | |
Kostniakomięsak, mięsak kościopochodny (łac. osteosarcoma) – grupa pierwotnych nowotworów złośliwych wywodzących się z prymitywnych komórek mezenchymatycznych klasyfikowanych do grupy pierwotnych wrzecionowatokomórkowych mięsaków tkanki kostnej produkujących osteoid[1][2].
Kostniakomięsak u ludzi jest stosunkowo rzadkim nowotworem[3], większość zachorowań dotyczy młodzieży i młodych dorosłych[4]. Kostniakomięsak występuje również u licznych gatunków zwierząt[5]. Nowotwór zwykle pojawia się w kościach długich w pobliżu płytki wzrostu, szczególnie w obrębie kości udowej, kości piszczelowej i kości ramiennej[1][6]. Nowotwór cechuje się dwoma szczytami zapadalności, pierwszym związanym z dorastaniem i skokiem wzrostowym oraz drugim przypadającym na wiek starszy[1][7]. Typowymi objawami klinicznymi kostniakomięsaka jest ból, obecność rozpierającej masy i obrzęk okolicy guza[7][8].
Pierwszym etapem diagnostyki kostniakomięsaka jest zdjęcie rentgenowskie w dwóch płaszczyznach; dopiero wówczas, gdy nie można wykluczyć mięsaka kości, konieczne jest wykonanie rezonansu magnetycznego[9]. Po postawieniu rozpoznania radiologicznego mięsaka kości konieczne jest przeprowadzenie biopsji guza[7], co pozwala dostarczyć materiału do badania histopatologicznego, które umożliwia ostateczne rozpoznanie kostniakomięsaka[10].
Stopień zaawansowania choroby warunkuje stosowane metody leczenia. W etapie choroby zlokalizowanej leczenie jest oparte o postępowanie chirurgiczne w skojarzeniu z chemioterapią okołooperacyjną. Leczenie chirurgiczne polega na radykalnej resekcji pierwotnego guza nowotworowego wraz z otaczającymi go i innymi potencjalnie zanieczyszczonymi komórkami nowotworowymi tkankami. Leczenie radykalne może polegać na operacji zachowującej kończynę albo wymagać jej amputacji[7]. Skojarzenie leczenia chirurgicznego z okołooperacyjną chemioterapią prowadzi do znacznego zwiększenia odsetku przeżyć pięcioletnich[11]. Leczenie okołooperacyjne polega na podaniu kilku kursów neoadiuwantowej chemioterapii mającej za zadanie ułatwienie lub umożliwienie leczenia chirurgicznego[12], następnie wykonania radykalnej operacji uzupełnionej kilkoma cyklami chemioterapii adiuwantowej[13][14]. W chorobie przerzutowej lub zaawansowanej miejscowo podstawową metodą leczenia jest chemioterapia oparta o doksorubicynę, cisplatynę i metotreksat w wysokich dawkach[12].
Objawy kliniczne

Typowym objawem klinicznym kostniakomięsaka jest ból, obecność rozpierającej masy i obrzęk okolicy guza[7][8]. Ból początkowo może mieć charakter przerywany, tępy i pojawiać się podczas aktywności fizycznej[7]. W późniejszym etapie ból stopniowo narasta w kolejnych miesiącach i występuje również w spoczynku, typowo jest silniejszy w nocy[7][15]. Często objawy bólowe są przypisywane znacznie częstszym tzw. bólom wzrostowym[7], które jednak nigdy nie mają charakteru ciągłego, a raczej przerywany i sporadyczny, pojawiając się kilka razy w ciągu miesiąca, zwykle w godzinach wieczornych lub nocnych, a ból jest słabo zlokalizowany i nie powoduje upośledzenia funkcji kończyny[16][17][18]. Objawy często są wiązane z wcześniejszym urazem danej okolicy, ale faktycznie uraz zwraca uwagę na chorą okolicę[7][15][19].
Obrzęk okolicy guza jest stwierdzany u 50% chorych[20], stwierdza się wówczas przekrwienie, zwiększoną ciepłotę i obrzęk tkanek nad zmianą[8]. Wysięk w stawie jest rzadki i sugeruje naciek stawu[20]. Zakres ruchomości kończyny może być zmniejszony, szczególnie w zaawansowanych przypadkach[8][20]. Rzadziej u 5–10% chorych choroba jest rozpoznawana z powodu złamania patologicznego[7][8][19]. W wyniku krwotoku z guza może dojść do bardzo szybkiego powiększenia się zmiany[21].
Objawy ogólnoustrojowe, na przykład gorączka i spadek masy ciała, rzadko występują w kostniakomięsaku[7]. W badaniu fizykalnym często stwierdza się bolesność palpacyjną w rzucie guza[7] i wyczuwalny nieprzesuwalny guz połączony z kością[22].
U 40% chorych obserwuje się podwyższoną aktywność fosfatazy alkalicznej (ALP), a u 30% chorych podwyższoną aktywność dehydrogenazy mleczanowej (LDH)[7].
Epidemiologia
Kostniakomięsak jest najczęstszym mięsakiem kości u ludzi[3]. Zapadalność na świecie na ten nowotwór nie różni się znacząco[6][23].
Zdecydowaną większość chorych (75%) stanowią osoby pomiędzy 15–25. rokiem życia[4]. Kostniakomięsak stanowi 10% nowotworów litych u osób w wieku nastoletnim[9]. Pod względem częstości stanowi ósmy nowotwór złośliwy wieku dziecięcego[1].
Przed 5. rokiem życia nowotwór występuje bardzo rzadko, następnie zapadalność stale wzrasta[24]. Szczyt zapadalności przypada na okres dojrzewania, u mężczyzn około 15–19. roku życia i u kobiet około 10–14. roku życia[23]. W grupie wiekowej poniżej 25. roku życia roczna zapadalność wynosi 3–5/1 000 000 u mężczyzn i 2–4/1 000 000 u kobiet[23]. Wcześniejszy wiek szczytu zapadalności u kobiet jest tłumaczony wcześniejszym wiekiem dojrzewania i wcześniejszym skokiem wzrostowym[7][25]. Wśród osób pomiędzy 25. i 59. rokiem życia zapadalność osiąga plateau i roczna zapadalność u mężczyzn wynosi 1,5–2,5/1 000 000 rocznie i u kobiet 0,5–2/1 000 000 rocznie[23]. Drugi szczyt zapadalności pojawia się u osób po 60. roku życia, w tej grupie wiekowej roczna zapadalność u mężczyzn wynosi 2,5–5/1 000 000 rocznie i u kobiet 1,5–4/1 000 000 rocznie[23]. W tej grupie wiekowej kostniakomięsak jest związany z wcześniejszą radioterapią, chorobą Pageta, choć u połowy chorych nie stwierdza się tych czynników przyczynowych[24]. Mężczyźni chorują 1,4-raza częściej niż kobiety[9], choć w grupie wiekowej poniżej 15. roku życia obserwuje się niewielką przewagę kobiet, a u chorych po 65. roku życia zapadalność u obu płci jest zbliżona[23].
W Polsce rocznie rozpoznaje się 60–100 przypadków kostniakomięsaka, co przekłada się na zapadalność na poziomie 2–3/1 000 000. 80% zachorowań przypada na 1. i 2. dekadę życia i blisko 20% na 6. i 7. dekadę[26].
Typy histopatologiczne
Kostniakomięsak jest nowotworem złośliwym charakteryzującym się bezpośrednim i pierwotnym wytwarzaniem przez komórki nowotworowego osteoidu lub niedojrzałej tkanki kostnej[4]. Nowotwory złośliwe kości w klasyfikacji WHO są dzielone następująco[27][28]:
- kostniakomięsak klasyczny (ang. conventional osteosarcoma, w ICD-O 9180/3 kostniakomięsak, bliżej nieokreślony ang. osteosarcoma, not otherwise specified),
- osteoblastyczny (ang. osteoblastic osteosarcoma, ICD-O 9180/3),
- chondroblastyczny (ang. chondroblastic osteosarcoma, ICD-O 9181/3),
- fibroblastyczny (ang. fibroblastic osteosarcoma, ICD-O 9182/3),
- kostniakomięsak teleangiektyczny (naczyniakowaty; ang. teleangiectatic osteosarcoma, ICD-O 9183/3),
- kostniakomięsak drobnokomórkowy (ang. small cell osteosarcoma, ICD-O 9185/3),
- kostniakomięsak przykostny (ang. parosteal osteosarcoma, ICD-O 9192/3),
- kostniakomięsak okostnowy (ang. periosteal osteosarcoma, ICD-O 9193/3),
- kostniakomięsak powierzchniowy o wysokim stopniu złośliwości (wewnątrzkorowy; ang. high grade surface osteosarcoma, ICD-O 9194/3),
- kostniakomięsak wewnątrzkostny o niskim stopniu złośliwości (śródkostny; ang. intraosseous low grade osteosarcoma; low grade central osteosarcoma, ICD-O 9187/3).
Kostniakomięsak pozaszkieletowy (pozakostny) (ang. extraskeletal osteosarcoma, ICD-O 9180/3) jest postacią zaliczaną do mięsaków tkanek miękkich[29].
Dodatkowo w klasyfikacji WHO kostniakomięsaki dzieli się na kostniakomięsaki centralne i powierzchniowe[4].
Kostniakomięsak może być zlokalizowany w obrębie dowolnej kości, jednak najczęściej występuje w przynasadach kości długich, szczególnie w dalszej części kości udowej, bliższej części kości piszczelowej i proksymalnej części kości ramiennej[30]. U ludzi kostniakomięsak w 80% jest zlokalizowany w obrębie kończyn, a w około 10% przypadków występuje w obrębie kości płaskich w tym żeber, miednicy, w 6–10% przypadków w obrębie czaszki i w 4% przypadków w obrębie kręgosłupa[31].
Lokalna progresja guza prowadzi do utraty funkcji kończyny[32]. Nowotwór szerzy się drogą naczyń krwionośnych, daje przerzuty odległe głównie do płuc, a w mniejszym stopniu do innych kości[33]. Przerzuty do lokalnych węzłów chłonnych są rzadko obserwowane[33], podobnie samo zajęcie szpiku jest rzadkie[33][34].
Charakterystyczne jest występowanie przerzutów skaczących (ang. skip metastases). Są to wtórne osobne ogniska nowotworowe w obrębie tej samej kości, niepozostające w ciągłości z guzem pierwotnym. Szacuje się, że występują u około 1% chorych. Są następstwem embolizacji naczyń zatokowych szpiku kostnego przez komórki nowotworowe, a zatem są to mikroprzerzuty, które nie przedostały się do krążenia głównego. Najczęściej występują w przypadku guzów o wysokiej złośliwości[35]. Możliwe jest występowanie przerzutów okołostawowych związanych z okołostawowymi zespoleniami żylnymi[35]. Bardzo rzadko stwierdzana jest wieloogniskowość choroby definiowana jako obecność co najmniej dwóch ognisk kostniakomięsaka bez obecności przerzutów w płucach. Mogą występować synchronicznie lub metachronicznie[36][37]. Szacuje się, że wieloogniskowość dotyczy około 1–3% chorych na kostniakomięsaka[36]. Jest kwestią sporną, czy zjawisko wieloogniskowości wynika z niezależnego pojawiania się ognisk nowotworowych, czy faktycznie są to przerzuty do kości, szczególnie w przypadku rozważania obecności synchronicznych ognisk[36][38]. Obecność wieloogniskowego kostniakomięsaka rozważa się w przypadku istnienia kilku ognisk o podobnej wielkości[38].
Lokalne nawroty są następstwem niecałkowitego wycięcia guza pierwotnego, zwykle pojawiają się w ciągu 24 miesięcy od leczenia radykalnego. Choroba nawrotowa cechuje się wysokim ryzykiem rozwoju przerzutów odległych i złym rokowaniem[39]. Nawrót choroby jest obserwowany u 30–40% chorych z pierwotnie zlokalizowaną chorobą[40]. U 10–20% chorych nowotwór jest rozpoznawany w stadium przerzutowym[30][33][40].
Kostniakomięsak klasyczny



Jest to śródszpikowy podtyp kostniakomięsaka o wysokiej złośliwości charakteryzujący się wytwarzaniem osseiny[41]. Jest najczęstszym typem histopatologicznym u ludzi i stanowi około 70–80% przypadków kostniakomięsaka[42][43].
Najczęstszą lokalizacją kostniakomięsaka klasycznego jest dalsza część kości udowej, bliższa część kości piszczelowej i proksymalna część kości ramiennej. Rzadko kostniakomięsak klasyczny jest zlokalizowany w żuchwie, kości szczękowej, miednicy, kręgosłupie i czaszce. Tendencję do obecności kostniakomięsaka poza kośćmi długimi obserwuje się u osób w starszym wieku. W 90% przypadków obejmuje przynasadę kości, tylko w 10% trzon, a zajęcie samej nasady jest niezwykle rzadkie[41].
Guzy zwykle osiągają duże rozmiary, zwykle powyżej 5 cm. Makroskopowo tworzą twardą lub mięsistą masę koloru szarobrązowego, część guzów jest bardziej sklerotyczna i przybiera kolor bardziej białożółty. Nowotwór często przekracza korę kości i nacieka tkanki miękkie[41].
Mikroskopowo guz jest zbudowany z wrzecionowatych komórek, choć faktycznie cechuje go tendencja do znacznego pleomorfizmu i anaplazji, przez co komórki mogą przybierać różne formy. W większości przypadków stwierdza się dwa i więcej typów komórek tworzących nowotwór. Również jądro komórkowe cechuje znaczny pleomorfizm. Nowotwór z definicji wytwarza osteoid, ale również może wytwarzać tkankę chrzęstną lub włóknistą. Ze względu na dominującą substancję pozakomórkową klasyczny kostniakomięsak może być dalej dzielony na postać osteoblastyczną, chondroblastyczną i fibroblastyczną[19][44].
Postać osteoblastyczna stanowi około połowy przypadków klasycznych kostniakomięsaków u ludzi[19][44]. Substancja pozakomórkowa tworzy cienką sieć pomiędzy pojedynczymi komórkami nowotworowymi. Nowotwory różnią się strukturą i gęstością substancji pozakomórkowej[45], która może być reprezentowana wyłącznie przez niezmineralizowaną osseinę do obecnych gęstych, zmineralizowanych beleczek kostnych. W części przypadków w utkaniu nowotworu przeważa substancja pozakomórkowa i komórki nowotworowe występują w niewielkiej ilości[44].
Postać chondroblastyczna stanowi około 25% przypadków klasycznych kostniakomięsaków u ludzi. W tym podtypie podstawową substancją pozakomórkową jest tkanka chrzęstna przypominającą chrząstkę szklistą przemieszaną z elementami niechrzęstnymi. Chrząstka jest zorganizowana w zraziki zawierające pojedyncze komórki z zaznaczoną anaplazją zlokalizowane w lukach pomiędzy substancją pozakomórkową. W obwodowej części guz staje się bogatokomórkowy[44]. Kostnina jest ubogo reprezentowana i może być obecna pomiędzy wrzecionowatymi komórkami a chrzęstnymi zrazikami[44][45].
Postać fibroblastyczna stanowi około 25% przypadków klasycznych kostniakomięsaków u ludzi. Jest to podtyp charakteryzujący się obecnością wrzecionowatych komórek z niewielką ilością substancji pozakomórkowej i obecność osteoidu jest bardzo uboga[46][47]. Utkanie może przypominać włókniakomięsaka lub niezróżnicowanego mięsaka pleomorficznego (malignant fibrous histiocytoma)[48].
Kostniakomięsak teleangiektyczny
Jest to podtyp kostniakomięsaka cechujący się obecnością dużych przestrzeni wypełnionych krwią z lub bez obecności przegród. Typowa jest obecność w RTG zmian litycznych bez tworzenia substancji pozakomórkowej oraz skłonność do występowania złamań patologicznych[48]. Jest to stosunkowo rzadki podtyp, stanowi około 3–4% przypadków kostniakomięsaka u ludzi[49][50][51].
Makroskopowo guz powoduje obszerną erozję prawidłowej tkanki kostnej, w obrazie utkania nowotworu dominuje obecność przestrzeni wypełnionych skrzepami krwi, makroskopowo nie stwierdza się obecności tworzenia osseiny (zmian wytwórczych). Torbiele przypominają tętniakowato poszerzone torbiele kostne, są wyłożone pleomoficznymi komórkami o wysokiej złośliwości[50][52]. Część guzów jest bardziej lita i torbiele stwierdza się mikroskopowo[52]. Ilość osteiodu jest różna, zwykle niewielka[53].
Kostniakomięsak drobnokomórkowy

Jest to podtyp histopatologiczny kostniakomięsaka zbudowany z małych komórek nowotworowych w różnym stopniu produkujący osteiod. Stanowi około 1,5% przypadków kostniakomięsaka u ludzi[54]. Makroskopowo nowotwór jest podobny do kostniakomięsaka klasycznego. Mikroskopowo budują go małe komórki, które mogą przybierać okrągły lub wrzecionowaty kształt[54]. Komórki mogą przypominać mięsaka Ewinga lub chłoniaka[54][55]. Komórki okrągłego kształtu mogą osiągać bardzo niewielkie rozmiary, zawierają niewielką ilość cytoplazmy, jądro jest okrągłe lub owalne. Komórki wrzecionowate cechują się krótkimi wrzecionowatymi jądrami, choć te mogą przyjmować okrągły kształt. W nowotworze zawsze stwierdza się obecność osteoidu[54].
Kostniakomięsak śródkostny
Jest to podtyp kostniakomięsaka wzrastający w obrębie jamy szpikowej, stanowi około 1–2% przypadków kostniakomięsaka u ludzi. Makroskopowo jest to szarobiały, lity guz o ziemistej teksturze rosnący w obrębie jamy szpikowej. Może niszczyć korę kości i penetrować w głąb tkanek miękkich[56].
Mikroskopowo guz jest zbudowany z ubogokomórkowego lub o umiarkowanej komórkowości fibroblastycznego zrębu ze zmienną ilością zrębu. Wrzecionowate komórki produkujące kolagen są ułożone w przeplatające się pęczki, które przenikają wcześniej istniejące beleczki kostne w jamie szpikowej. Komórki nowotworowe zazwyczaj wykazują nieznaczną atypię[56][57]. Nowotwór charakteryzuje się zmiennością wzorca produkcji osteoidu, w części przypadków utkanie beleczek kostnych może przypominać dysplazję włóknistą kości. W 15–20% przypadków kostniakomięsak ulega progresji do niezróżnicowanego mięsaka wrzecionowatokomórkowego o wysokim stopniu złośliwości[58].
Kostniakomięsak przykostny
Jest to kostniakomięsak o niskiej złośliwości histologicznej wzrastający na powierzchni kości. Stanowi około 4% przypadków kostniakomięsaka u ludzi[59]. Makroskopowo tworzy twardą zmianę połączoną z niżej leżącą korą kości, brzegi zmiany mogą być bardziej miękkie i może być widoczna inwazja w obręb tkanek miękkich. W co czwartym przypadku obecna jest inwazja szpiku kostnego. Guz wykazuje tendencję do owijania się wokół zajętej kości. Makroskopowo może być widoczna tkanka chrzęstna zorganizowana w guzki w obrębie utkania nowotworu[59].
Mikroskopowo guz jest zbudowany z dobrze uformowanych beleczek kostnych z ubogokomórkowym zrębem pomiędzy nimi[59][60]. Komórki wrzecionowate wykazują niewielką atypię[60], a w 20% przypadków zrąb zawiera więcej komórek, które wykazują umiarkowaną atypię[59]. W obrzeżach zmiany komórki wrzecionowate wykazują bardziej nasiloną proliferację bez produkcji substancji pozakomórkowej i postępują w obręb tkanek miękkich[60].
W połowie guzów widoczne jest tworzenie tkanki chrzęstnej[59]. Tkanka chrzęstna może być zorganizowana w bogatokomórkowe guzki w substancji pozakomórkowej nowotworu lub tworzyć „czapeczkę” pokrywającą guz upodabniając go do chrzęstniakokostniaka[61]. W 15% przypadków guza są obecne obszary odróżnicowania niezróżnicowanego mięsaka wrzecionowatokomórkowego o wysokiej złośliwości[62][63].
Kostniakomięsak okostnowy
Jest to podtyp kostniakomięsaka o pośrednim stopniu złośliwości wzrastający na powierzchni trzonu kości długich. Stanowi około 2% przypadków kostniakomięsaka u ludzi[64]. Makroskopowo guz wyrasta z powierzchni kości, może obejmować jej część lub cały obwód, przyjmuje płatowaty wygląd, wyglądem może przypominać chrząstkę[65].
Mikroskopowo kostniakomięsak okostnowy przypomina umiarkowanie zróżnicowanego chrzęstniakomięsaka[66][67]. W utkaniu nowotworu przeważa komponent chrzęstny, wykazuje on różny poziom zróżnicowania i atypii[66][67], jest zorganizowany w guzki[68][69]. Osteoid jest bardziej zaznaczony na obrzeżach guza, gdzie są obecne komórki wrzecionowate ułożone w nieregularne pęczki[66][67], dojrzałe beleczki kostne są nieobecne. Guz wykazuje niewielką tendencję do inwazji tkanek miękkich[69].
Kostniakomięsak powierzchniowy o wysokim stopniu złośliwości
Jest to podtyp kostniakomięsaka o wysokim stopniu złośliwości tworzącym osseinę. Stanowi poniżej 1% przypadków kostniakomięsaka u ludzi. Nowotwór wzrasta na powierzchni kości i powoduje niszczenie niżej leżącej kory. Powierzchnia guza jest zrazikowa, a konsystencja jest różna w zależności od obecności komponentu osteoblastycznego, chondroblastycznego i fibroblastycznego, jednak niemal każdy guz zawiera miękkie obszary, co ułatwia jego odróżnienie od innych podtypów[70].
Mikroskopowo stwierdza się obszary wrzecionowatych komórek o znacznej atypii i zróżnicowanej ilości tworzonego osteoidu[69]. Wyróżnia się podtypy o różnicowaniu osteoblastycznym, chondroblastycznym i fibroblastycznym[70].
Kostniakomięsak pozaszkieletowy (pozakostny)
Jest to nowotwór zaliczany do mięsaków tkanek miękkich w kategorii nowotworów z tkanki chrzęstnej i kostnej (ang. chondro-osseus tumors)[29]. Jest to rzadki nowotwór z obecnymi komórkami o morfologii osteoblastów tworzących osteoid lub niedojrzałą tkankę kostną[71]. Postać pozakostna z definicji jest zlokalizowana w obrębie tkanek miękkich i nie może być związana z kością lub okostną[72]. Stanowi około 2–5% przypadków kostniakomięsaka u ludzi[71][72][73].
Zwykle są to nowotwory o wysokiej złośliwości histologicznej i agresywnym przebiegu klinicznym. U ludzi najczęściej pojawia się w 5.–7. dekadzie życia, w 10% w powierzchownych tkankach miękkich[71]. Zwykle jest zlokalizowany w obrębie głębokich tkanek proksymalnych części kończyn, przede wszystkim uda[74]. Rzadziej występuje w obrębie przestrzeni zaotrzewnowej[72].
Makroskopowo są to dobrze odgraniczone guzy koloru białego do jasnobrązowego, jednak faktycznie mikroskopowo wykazują naciekanie sąsiednich tkanek. Mogą być obecne ogniska krwotoczne lub martwicy[71]. Mikroskopowo jest to bogatokomórkowy guz o znacznym pleomorfizmie cytologicznym[74]. Postać pozaszkieletowa może prezentować wszystkie najważniejsze warianty kostniakomięsaka szkieletowego, w tym typ fibroblastyczny, chondroblastyczny, naczyniakowaty i drobnokomórkowy. Atypowe komórki mają kształt wrzecionowaty lub wielościenny. Wspólną cechą dla wszystkich wariantów jest obecność nowotworowego osteoidu lub niedojrzałej tkanki kostnej ściśle związanego z komórkami nowotworowymi, może występować tkanka chrzęstna[71].
Patogeneza
Etiologia choroby pozostaje nieznana[4][75]. Przyjmuje się, że choroba jest następstwem podatności genetycznej i interakcji czynników środowiskowych[76][77]. Ze względu na stosunkową rzadkość nowotworu, badania czynników ryzyka choroby są ograniczone do niewielkich grup chorych[78].
Rola czynników środowiskowych i endogennych
- Skok wzrostowy
Kostniakomięsak jest związany ze skokiem wzrostowym w okresie dojrzewania i związanym z nim szybkim wzrostem kości. Najczęściej pojawia się pod koniec wzrostu kości, szczególnie w obrębie kości długich. Ponadto pierwszy szczyt zapadalności pojawia się we wcześniejszym wieku u kobiet niż u mężczyzn, co ma związek z niższym wiekiem wejścia w okres dojrzewania i przyspieszenia wzrostu u dziewczynek niż u chłopców[79]. W dużym badaniu epidemiologicznym stwierdzono, że kostniakomięsak częściej występuje u wyższych osób[7][80][81]. Podobną zależność wykazano u psów, u których częściej stwierdza się kostniakomięsaka u przedstawicieli większych ras tych zwierząt[82][83].
- Choroba Pageta
Choroba Pageta jest czynnikiem ryzyka zachorowania na kostniakomięsaka, szczególnie u osób po 40. roku życia, gdzie choroba może stanowić przyczynę 20% przypadków kostniakomięsaka u ludzi[76]. Szacuje się, że u około 1% chorych z chorobą Pageta rozwinie się kostniakomięsak[84]. Bardzo rzadko kostniakomięsak rozwija się na bazie chrzęstniakokostniaka (osteochondroma), chrzęstniaka śródkostnego, kostniakochrzęstniaka, choroby Olliera, dysplazji włóknistej kości i przewlekłego zapalenia kości[76].
- Promieniowanie jonizujące i leczenie przeciwnowotworowe
Promieniowanie jonizujące jest związane z około 3% przypadków kostniakomięsaka u ludzi[85]. Obserwuje się zwiększone ryzyko zachorowania u chorych poddanych wysokodawkowej radioterapii kości, szczególnie chorych leczonych z powodu mięsaka Ewinga[85][86][87]. Prawdopodobnie leki alkilujące oraz antracykliny mogą zwiększać ryzyko zachorowania na kostniakomięsaka[88][89].
- Teoria wirusowa
Nie ma dowodów naukowych łączących zachorowania wirusowe ze zwiększonym ryzykiem zachorowania na kostniakomięsaka[90].
Podłoże cytogenetyczne
Na poziomie cytogenetycznym w nowotworzeniu kostniakomięsaka kluczowa jest aktywacja kilku protoonkogenów, wyłączenie kilku genów supresorowych oraz genów mających znaczenie dla utrzymania stabilności genomowej[91]. Nie rozpoznano pojedynczej powtarzalnej zmiany cytogenetycznej, która mogłaby być odpowiedzialna za patogenezę tego nowotworu. Zmiany cytogenetyczne związane z kostniakomięsakiem dzieli się na dwie grupy: zmiany związane z kontrolą proliferacji oraz zmiany związane z kontrolą cyklu komórkowego i naprawy DNA[77].
- Szlak Wnt/β-katenina
Fizjologicznie szlak Wnt/β-katenina pełni ważną funkcję w powstaniu i rozwoju tkanki kostnej, a także w regulacji jej metabolizmu i masy[92]. Wiele badań wskazuje, że szlak Wnt/β-katenina odgrywa istotną rolę w patogenezie kostniakomięsaka[93]. W jednym badaniu w 70% przypadków ludzkiego kostniakomięsaka stwierdzono zaburzenia tego szlaku[94].
Do aktywacji szlaku Wnt dochodzi poprzez zwiększenie sekrecji białek Wnt, zwiększenia ekspresji receptorów Wnt lub zmniejszenia ekspresji inhibitorów[92]. Szlak jest zapoczątkowywany przez związanie białka Wnt z receptorem frizzled i koreceptorem LRP5/6, w wyniku czego za pośrednictwem kilku białek dochodzi do aktywacji kilku efektorów tego szlaku, w tym β-kateniny. Nagromadzenie β-kateniny w cytoplazmie i jej translokacja do jądra komórkowego, a następnie jej związanie z LEF/TCF prowadzi do aktywacji wielu onkogenów, w tym genu c-MYC, cycliny D1, metaloproteinaz i c-MET[95]. Aktywacja TCF jest odpowiedzialna za hamowanie proapoptycznego syndekanu-2, który jest istotny w regulacji apoptozy, a w konsekwencji sprzyja oporności na cytostatyki[95][96][97]. Z kolei metaloproteinazy macierzy zewnątrzkomórkowej (MMP) poprzez rozkład macierzy pozakomórkowej sprzyjają inwazji i migracji komórek nowotworowych[95][98].
- Szlak PI3K/AKT
Szlak PI3K/AKT odpowiada za wiele procesów komórkowych zarówno fizjologicznych, jak i tych patologicznych. Odgrywa istotną rolę w regulacji wzrostu komórki, oporności na apoptozę, zdolności do migracji, zdolności do wytwarzania przerzutów[95]. W większości przypadków zlokalizowanego kostniakomięsaka i niemal we wszystkich przypadkach zaawansowanego kostniakomięsaka jest obserwowana deregulacja szlaku PI3K/AKT, co sugeruje, że zaburzenia w obrębie tego szlaku mogą warunkować etap progresji tego nowotworu[99]. AKT za pośrednictwem hamowania GSK-3β nasila ekspresję onkoproteiny MYC i ekspresję CDK4. Poprzez hamowanie FOXO1 hamuje ekspresję p27 i p21 nasilając ekspresję CDK, co wpływa na promocję wzrostu i podziału komórek. Za pośrednictwem MDM2 szlak PI3K/AKT hamuje białko p53 pełniące funkcję białka supresorowego. Poprzez regulację w górę Bcl-xl, Bcl-2 i Mcl1 oraz regulację w dół Bad, Bax i p53 dochodzi do inhibicji apoptozy[99]. Zablokowanie szlaku PI3K/AKT powoduje spadek ekspresji genów metoproteinaz MMP-2 i MMP-9, które pełnią istotną rolę w rozkładzie macierzy pozakomórkowej, wpływając na zdolność do migracji i inwazji komórek nowotworowych[99][100].
- p53
Białko p53 pełni funkcję supresora nowotworowego wykazując zdolność do aktywacji naprawy DNA lub indukcji apoptozy komórki, jego inaktywacja jest stwierdzana w 10–39% przypadków kostniakomięsaka. W zespole Li-Fraumeni, gdzie występuje wrodzona mutacja genu TP53, obserwuje się zwiększone ryzyko zachorowania również na kostniakomięsaka[75][101].
- RB1
Dezaktywacja genu supresorowego RB1 w wyniku mutacji lub delecji jest stwierdzana w około 50% przypadków kostniakomięsaka klasycznego. Gen RB1 koduje białko RB, które w przypadku uszkodzenia DNA, poprzez związanie i zablokowanie czynników transkrypcyjnych należących do rodziny E2F, powoduje zablokowanie przejścia cyklu komórkowego z fazy G1 i przejściu w fazę S. Mutacje genu w linii zarodkowej znacznie zwiększają ryzyko zachorowania na kostniakomięsaka[75].
- CDK
Kinazy zależne od cyklin (CDK) są grupą kinaz pełniących funkcję regulatorową cyklu komórkowego. Progresja cyklu komórkowego wymaga aktywności CDK4, CDK6, CDK2 i CDK1. CDK4/6 z cyklinami D1, D2 lub D3 oraz CDK2 inaktywują RB i promują przejście do fazy S. Z kolei CDK2 i CDK1 z odpowiednimi cyklinami promują przejście z fazy S do dalszych etapów cyklu komórkowego[102].
W 10% przypadków stwierdza się amplifikację genu CDK4 i w 40% genu PRIM1 wpływających na kontrolę przejścia z fazy G1 do S, jednak rola zwiększenia liczby ich kopii w nowotworzeniu kostniakomięsaka jest niejasna[75]. Stwierdzono także mutacje genów supresorowych p16 (CDKN2A), p14 (ARF) i p15 (CDKN2B) wpływających na inaktywację CDK[103][75]. Również szlak PI3K/AKT wpływa na ekspresję CDK[99].
- c-MYC
W kostniakomięsaku stwierdza się niestabilność chromosomalną chromosomu 8q. Chromosom zawiera protoonkogen c-MYC w locus 8q24.21, którego zwiększenie liczby kopii stwierdza się w 14–67% przypadków tego nowotworu[75]. Z drugiej strony stwierdzono zwiększenie kopii również innych regionów tego chromosomu, dlatego prawdopodobnie również inne onkogeny zlokalizowane w 8q mogą mieć rolę w patogenezie choroby[104][75]. MYC jest czynnikiem transkrypcyjnym odpowiedzialnym za wiele procesów komórkowych, w przypadku nadekspresji pełni funkcję onkogenu stymulującego wzrost komórek i zdolność do podziałów[101]. Prawdopodobnie poprzez szlak MEK-ERK wpływa na zdolność do inwazji komórek nowotworowych kostniakomięsaka[105]. Amplifikacja genu c-MYC jest stwierdzana w 7–14% przypadków kostniakomięsaka u ludzi[99].
- TGF-β
Transformujący czynnik wzrostu beta (TGF-β) jest czynnikiem wzrostu wpływającym na proliferację, różnicowanie, apoptozę i produkcję macierzy pozakomórkowej[101]. W kostniakomięsaku o wysokim stopniu złośliwości zaobserwowano wyższą ekspresję TGF-β w porównaniu do kostniakomięsaka o niskim stopniu złośliwości[106]. Prawdopodobnie tworzy pętle dodatniego sprzężenia zwrotnego, nasilając proliferację i przebudowę macierzy pozakomórkowej, sprzyjając progresji, migracji i inwazji nowotworu[107][108].
- GSK-3β i NF-κB
Kinaza syntazy glikogenu 3β jest kinazą białkową pełniącą w przypadku kostniakomięsaka rolę onkoproteiny (w niektórych nowotworach złośliwych odgrywa rolę supresora nowotworowego)[95]. Niektóre badania wskazują na krytyczną rolę GSK-3β w przetrwaniu komórek nowotworowych kostniakomięsaka, które po farmakologicznym zablokowaniu GSK-3β ulegają apoptozie[109]. Jednocześnie badania wskazują, że blokada GSK-3β wpływa na zablokowanie szlaku NF-κB[110].
- RECQL4
Ze zwiększonym ryzykiem kostniakomięsaka jest związana mutacja genu RECQL4 związana z zespołem Rothmunda-Thomsona, ale przypadki sporadycznej mutacji nie są częste i mutacja występuje w 5% przypadków kostniakomięsaka[111]. Gen RECQL4 koduje białko należące do rodziny helikaz RecQ istotnych dla regulacji naprawy DNA i kontroli replikacji wpływając na stabilność chromosomalną[112][113]. Również zespół Blooma[114] oraz zespół Wernera[115] są uwarunkowane mutacjami helikaz rodziny RecQ i są związane ze zwiększonym ryzykiem rozwinięcia kostniakomięsaka[75].
- 6p12-p21
W 16–75% przypadków kostniakomięsaka stwierdza się amplifikację regionu 6p12-p21[75]. W regionie 6p12-p21 jest zlokalizowany gen czynnika transkrypcyjnego E2F3, którego zwiększona ekspresja sprzyja akumulacji uszkodzeń materiału genetycznego oraz nasila proliferację komórek nowotworowych[75][116][117].
W regionie 6p12-p21 zlokalizowane są również inne istotne geny dla procesu nowotworzenia: protoonkogen PIM1[118], gen VEGF-A odpowiedzialny za neoangiogenezę[119], gen CCND3 kodujący cyklinę D3[75][120], gen CDC5L wpływający na regulację cyklu komórkowego[121] i gen RUNX2 wpływający na osteogenezę[122]. Utrata heterozygotyczności w locus 10q26 wpływa na utratę genu BUB3, który jest ważny dla utrzymania stabilności genonowej[75][123].
Rozpoznanie
Zdjęcie rentgenowskie w dwóch płaszczyznach jest pierwszym etapem diagnostyki kostniakomięsaka. Jeśli na tym etapie nie udaje się wykluczyć rozpoznania mięsaka kości, zaleca się wykonanie rezonansu magnetycznego. Tomografia komputerowa znajduje zastosowanie w przypadkach wątpliwych, gdy występuje konieczność potwierdzenia obecności kalcyfikacji zmiany, osteolizy lub osteosklerozy[9].
Po wykonaniu badań obrazowych stawia się kliniczne podejrzenie mięsaka kości i konieczne jest przeprowadzenie biopsji guza, najlepiej w ośrodku doświadczonym w leczeniu mięsaków kości[7]. Preferuje się biopsję otwartą, choć rozpoznanie bywa stawiane po uzyskaniu materiału z biopsji gruboigłowej lub trepanobiopsji[124]. Ostateczne rozpoznanie jest stawiane na podstawie badania histopatologicznego materiału pobranego na drodze biopsji operacyjnej lub oligobiopsji[10].
Zdjęcie radiologiczne

Zdjęcie rentgenowskie w dwóch płaszczyznach jest podstawowym badaniem u chorych z podejrzeniem mięsaka kości[9][125]. Ocenie podlega lokalizacja zmiany, jej wielkość, obecność osteolizy, osteosklerozy oraz obraz odczynów okostnowych[125].
Radiologicznie w zależności od odczynu kostnego wyróżnia się postać osteolityczną i osteosklerotyczną[126]. Osteoliza polega na bezodczynowym niszczeniu kości z następczym przerwaniem jej warstwy korowej. Z kolei odczyn okostnej powoduje nawarstwienie kory, która ulega przerwaniu w miejscu infiltracji guza w głąb tkanek miękkich, dając objaw radiologiczny nazywany trójkątem Codmana lub mankietem Codmana związany z odwarstwieniem okostnej od kości[126]. Jednak objaw nie jest swoisty wyłącznie dla kostniakomięsaka[7]. Mogą być stwierdzane prostopadłe w stosunku do powierzchni kości igiełki lub liczne drobne przejaśnienia bez odczynu kostnego (tzw. kość wygryziona przez mole). W postaci osteosklerotycznej nowotwór może być obserwowany jako zagęszczenia struktury kości bez wyraźnego odgraniczenia od prawidłowej tkanki kostnej[126].
Tomografia komputerowa
Tomografia komputerowa (TK) jest wykorzystywana do oceny zaawansowania miejscowego guza, jednak do oceny naciekania tkanek miękkich, nacieku szpiku oraz przerzutów do szpiku kostnego lepszym badaniem jest rezonans magnetyczny. Do oceny obecności przerzutów w płucach tomografia komputerowa jest badaniem bardziej czułym od RTG klatki piersiowej[7].
Rezonans magnetyczny
Rezonans magnetyczny (MRI) jest najdokładniejszą metodą do oceny zaawansowania miejscowego guza[28][127][128]. Badanie powinno obejmować całą kość oraz stawy ponad i pod lokalizacją guza pierwotnego. Badanie pozwala rozpoznać obecność infiltracji nowotworu w obręb tkanek miękkich, jamy szpikowej, obecność nacieku stawów, płytki wzrostu oraz struktur naczyniowo-nerwowych[28].
PET
Pozytonowa tomografia emisyjna (PET) w połączeniu z TK lub MRI pozwala wykrywać miejsca o wzmożonej aktywności metabolicznej, które mogą odpowiadać ogniskom choroby nowotworowej. Metoda znajduje zastosowanie w ocenie zaawansowania choroby, wyboru najlepszej lokalizacji do wykonania biopsji, oceny odpowiedzi na leczenie, wykrycia nawrotu choroby, planowaniu radioterapii oraz diagnostyki różnicowej zmian złośliwych i niezłośliwych[28].
Scyntygrafia
Scyntygrafia za pomocą izotopu Tc99 MDP jest wykorzystywana do wykrywania przerzutów do kości kostniakomięsaka[7][28].
Biopsja i badanie histopatologiczne
.jpg)
Biopsja umożliwia pobranie materiału do badania histopatologicznego i ustalenie ostatecznego rozpoznania kostniakomięsaka[28]. Jest wykonywana po wykonaniu badań obrazowych i postawieniu radiologicznego (klinicznego) podejrzenia kostniakomięsaka[7]. Powinna być wykonana w ośrodku doświadczonym w leczeniu mięsaków kości[124]. Miejsce wykonania biopsji jest wybierane na podstawie badań obrazowych i scyntygrafii kości oraz zasadą najkrótszego odcinka pomiędzy guzem a skórą[124]. Miejsce pobrania materiału powinno znajdować się daleko od pęczków naczyniowo-nerwowych, a następnie miejsce biopsji powinno zostać wycięte w ramach ostatecznego leczenia[10]. Konieczne jest uzyskanie żywego utkania tkanki nowotworowej oraz zrębu nowotworu[124].
Biopsja może być wykonana metodą otwartą lub gruboigłowo[28]. Biopsja otwarta jest podstawową metodą biopsji w przypadku mięsaków kości, jest przeprowadzana w znieczuleniu ogólnym i polega na wycięciu całego guza lub reprezentatywnego fragmentu guza[129]. Jednak otwarta biopsja wiąże się z większym ryzykiem rozsiania komórek nowotworowych w prawidłowych tkankach[28].
Część ośrodków ma odpowiednie doświadczenie do oparcia diagnostyki i leczenia o biopsję gruboigłową lub trepanobiopsję[124]. Biopsja gruboigłowa pobrana z kilku miejsc guza może być wystarczającą metodą do uzyskania reprezentatywnych bioptatów[28]. Metoda pozwala ustalić rozpoznanie u 88–96% chorych[124] i w doświadczonych ośrodkach stanowi alternatywę dla biopsji metodą otwartą[9]. Biopsja gruboigłowa wiąże się z mniejszym ryzykiem rozsiewu komórek nowotworowych w zdrowych tkankach niż metoda otwarta. W celu zwiększenia skuteczności diagnostycznej biopsja może być wykonywana pod kontrolą USG lub TK[28]. Miejsce wykonania biopsji powinno być oznaczone nacięciem lub tatuażem[130]. Biopsja cienkoigłowa pozwala jedynie na wykonanie badania cytologicznego i nie ma większej roli w diagnostyce mięsaków kości[28]. Ostateczne rozpoznanie jest stawiane na podstawie badania histopatologicznego materiału pobranego na drodze biopsji operacyjnej lub oligobiopsji[10].
Zaawansowanie kliniczne
Zaawansowanie kostniakomięsaka jest oceniane w klasyfikacji TNM dla mięsaków kości.
| Guz pierwotny – cecha T | |
| Tx | nie można ocenić guza pierwotnego |
| T0 | nie stwierdza się obecności guza pierwotnego |
| T1 | guz mniejszy lub równy 8 cm w największym wymiarze |
| T2 | guz większy niż 8 cm w największym wymiarze |
| T3 | oddzielne ogniska nowotworowe w obrębie pierwotnej kości – przerzuty skaczące (skip metastases) |
| Zajęcie okolicznych węzłów chłonnych – cecha N | |
| Nx | nie można ocenić regionalnych węzłów chłonnych |
| N0 | nie stwierdza się przerzutów w regionalnych węzłach chłonnych |
| N1 | obecność przerzutów w regionalnych węzłach chłonnych |
| Przerzuty odległe – cecha M | |
| Mx | nie można ocenić regionalnych węzłów chłonnych |
| M0 | nie stwierdza się przerzutów odległych |
| M1 | obecne przerzuty odległe |
| M1a | przerzuty do płuc |
| M1b | przerzuty do innych narządów odległych |
| Stopień złośliwości histologicznej – cecha G | |
| Gx | nie można ocenić stopnia złośliwości histologicznej |
| G1 | dobrze zróżnicowany nowotwór – niski stopień złośliwości histologicznej |
| G2 | pośrednio zróżnicowany nowotwór – niski stopień złośliwości histologicznej |
| G3 | źle zróżnicowany nowotwór – wysoki stopień złośliwości histologicznej |
| G4 | odróżnicowany nowotwór – wysoki stopień złośliwości histologicznej |
| Stopień zaawansowania | Cecha T | Cecha N | Cecha M | Cecha G |
| IA | T1 | N0 | M0 | G1 |
| T1 | N0 | M0 | G2 | |
| IB | T2 | N0 | M0 | G1 |
| T2 | N0 | M0 | G2 | |
| T3 | N0 | M0 | G1 | |
| T3 | N0 | M0 | G2 | |
| IIA | T1 | N0 | M0 | G3 |
| T1 | N0 | M0 | G4 | |
| T2 | N0 | M0 | G3 | |
| T2 | N0 | M0 | G4 | |
| IIB | T2 | N0 | M0 | G3 |
| T2 | N0 | M0 | G4 | |
| III | T3 | N0 | M0 | G3 |
| T3 | N0 | M0 | G4 | |
| IVA | każde T | N0 | M1a | każde G |
| IVB | każde T | każde N | M1b | każde G |
| każde T | N1 | każde M | każde G |
Leczenie
Stopień zaawansowania choroby warunkuje stosowane metody leczenia. W etapie choroby zlokalizowanej leczenie jest oparte o leczenie skojarzone obejmujące radykalny onkologicznie zabieg chirurgiczny w skojarzeniu z chemioterapią okołooperacyjną. Leczenie chirurgiczne może polegać na operacji zachowującej kończynę albo wymagać jej amputacji[7]. Leczenie okołooperacyjne polega na podaniu kilku kursów neoadiuwantowej chemioterapii poprzedzającej zabieg oraz kilku cykli chemioterapii adiuwantowej uzupełniającej zabieg[13][14]. W chorobie przerzutowej lub zaawansowanej miejscowo podstawową metodą leczenia jest chemioterapia oparta o doksorubicynę, cisplatynę i metotreksat w wysokich dawkach[12].
Leczenie chirurgiczne choroby zlokalizowanej
Całkowite chirurgiczne usunięcie zmiany nowotworowej jest kluczowe do osiągnięcia wyleczenia choroby[135]. Leczenie chirurgiczne polega na całkowitej resekcji pierwotnego guza nowotworowego wraz z otaczającymi go tkankami, miejscem wykonania biopsji, miejscami przejścia drenów i innych potencjalnie zanieczyszczonych komórkami nowotworowymi tkanek[7]. Leczenie radykalne może wymagać amputacji kończyny, jednak często możliwe jest leczenie zachowujące kończynę[7][135].
Szerokie wycięcie guza (wewnątrzprzedziałowe) oznacza usunięcie guza w całości (en bloc), strefy reaktywnej wraz z marginesem zdrowych otaczających tkanek bez przekraczania pojedynczego przedziału mięśniowego. Wycięcie radykalne polega na wycięciu całej zajętej struktury z obecnym guzem, a płaszczyzna preparacji przebiega poza powięzią lub granicami kostnymi[136].
- Leczenie zachowujące kończynę
W 80–90% przypadków możliwa jest chirurgiczna resekcja z odpowiednią rekonstrukcją pozwalająca na zachowanie funkcjonalnej kończyny[135]. Leczenie wymaga radykalnej onkologicznie resekcji guza z uzyskaniem marginesu resekcji wolnego od nacieku nowotworowego zarówno w obrębie kości, jak i tkanek miękkich. Kluczowe dla skuteczności leczenia jest zapobiegnięcie wznowie miejscowej[136]. Niezbędne jest zachowanie 3–4 cm marginesu resekcji tkanki kostnej, który jest wyznaczany w badaniach obrazowych (TK lub MRI), a także resekcja sąsiedniego stawu i torebki stawowej[137]. Następnie konieczne jest odtworzenie ubytku kości, który zwykle mierzy 15–20 cm[136]. Techniki rekonstrukcji różnią się w zależności od resekcji, stosuje się endoprotezy kostne lub przeszczepy kostne[137]. Wymagana jest rekonstrukcja ubytku tkanek miękkich, w tym mięśni. Wykonuje się przemieszczenie płatów mięśniowych lub skórno-mięśniowych[136].
Podstawowym warunkiem przeprowadzenia operacji zachowujących kończynę jest możliwość uzyskania lepszego efektu czynnościowego niż amputacja i protezowanie zewnętrzne bez pogorszenia jakości życia[138]. Operacja oszczędzająca musi prowadzić do zachowania w kończynie dolnej funkcji podporowej i możliwości chodzenia, w kończynie górnej funkcji chwytnej ręki, a także nieobecności bólu, zachowania czucia głębokiego i powierzchniowego. Zabieg oszczędzający może być planowany wyłącznie u chorych, u których uzyskano stabilizację (SD) lub częściową odpowiedź (PR) na chemioterapię[139][138]. Przeciwwskazaniem do zabiegu oszczędzającego jest zajęcie struktur naczyniowo-nerwowych. Choć możliwe jest wstawienie protezy naczyniowej, to jednak ze względu na znaczne prawdopodobieństwo zajęcia przebiegających w pobliżu nerwów, wykonanie radykalnego onkologicznie zabiegu chirurgicznego staje się mało prawdopodobne[137].
Względnym przeciwwskazaniem do operacji zachowującej kończynę jest złamanie patologiczne[139]. Jest to związane ze zwiększonym ryzykiem wznowy miejscowej spowodowanym rozsiewem komórek nowotworowych w obrębie utworzonego podczas złamania krwiaka[137].
Kostniakomięsak w proksymalnej części kości ramiennej zwykle wymaga przeprowadzenia pozastawowej resekcji obejmującej usunięcie 15–20 cm kości ramiennej, usunięcia stawu barkowego, mięśnia naramiennego, pierścienia rotatorów i części mięśnia dwugłowego ramienia oraz mięśnia trójgłowego ramienia. Z kolei kostniakomięsak w dystalnej części kości udowej i proksymalnej części kości piszczelowej zwykle wymaga usunięcia 15–20 cm dystalnej części kości udowej i proksymalnej części kości piszczelowej wraz z przylegającymi fragmentami mięśnia czworogłowego. Duże guzy mogą wymagać usunięcia całego mięśnia czworogłowego[140].
Trudności powoduje niedojrzałość kostna i niezakończony wzrost kostny. Konieczność usunięcia przynasad w dotkniętej chorobą kończynie przyczynia się do zaburzenia jej wzrostu, który w 70% zależy od płytek wzrostu położonych w przynasadach[141]. W konsekwencji dochodzi do powstania asymetrii długości wobec zdrowej kończyny, w tym również różnej wysokości stawów kolanowych. Przewidywalna różnica długości kończyn nie powinna przekraczać 6–8 cm. Rozwiązaniem mogą być wydłużalne protezy („rosnąca proteza”)[137].
W badaniu na 227 chorych z kostniakomięsakiem leczonych amputacją lub operacją zachowującą kończynę, stwierdzono podobny odsetek nawrotów miejscowych wynoszący odpowiednio 8% i 11% oraz nie stwierdzono różnic w osiąganym przeżyciu całkowitym[142][143]. W innym badaniu na 540 chorych leczonych operacją zachowującą kończynę stwierdzono lokalny nawrót choroby u 8% ze słabą odpowiedzią histologiczną na leczenie neoadiuwantowe i u 3% z dobrą odpowiedzią histologiczną[135][143]. Nawroty lokalne cechują się złym rokowaniem[135].
- Amputacja
Amputacja jest metodą leczenia kostniakomięsaka u chorych niekwalifikujących się do leczenia zachowującego kończynę oraz u chorych, u których zabieg oszczędzający nie byłby odpowiedni[144]. Dawniej amputacja kończyny była uważana za jedyną metodę leczenia mięsaków w obrębie kończyny dolnej[145]. Obecnie tylko około 10–15% chorych wymaga tej metody leczenia. Zwykle są konieczne wysokie amputacje, które w przeciwieństwie do amputacji z przyczyn nienowotworowych są zabiegami trudnymi technicznie[144].
- Plastyka rotacyjna
Plastyka rotacyjna jest operacją zastąpienia stawu kolanowego stawem skokowym. Zabieg polega na resekcji dalszej części kości udowej i bliższej kości piszczelowej, a następnie wykonania zespolenia kikuta kości udowej z pozostałą dalszą częścią kości piszczelowej z zachowaną stopą z wykonaniem rotacji podudzia o 180°. Chory po zabiegu wymaga zaopatrzenia protetycznego funkcjonalnie nieobecnego (przemieszczonego w miejsce dalszej części amputowanego uda) podudzia ze stawem skokowym (pełniącego po operacji funkcję stawu kolanowego)[145][146][147]. Operacja może być wykonana, gdy nie ma konieczności amputacji proksymalnej części kości piszczelowej[148]. Warunkiem operacji jest zachowanie nerwu kulszowego[148]. Zabieg jest wykonywany u chorych z bardzo niedojrzałym układem kostno-stawowym (w wieku 8–10 lat), u starszych chorych niekwalifikujących się do operacji zachowania kończyny i jako operacja ratująca kończynę przy przewlekle zakażonych implantach[148].
Leczenie okołooperacyjne
| Odpowiedź | Badanie histopatologiczne |
| Całkowita odpowiedź | Nieobecność żywego utkania nowotworowego |
| Dobra odpowiedź | 90–99% martwicy nowotworu |
| Słaba odpowiedź | 60–89% martwicy nowotworu |
| Zła odpowiedź | <60% martwicy nowotworu |
Leczenie skojarzone jest kluczowym elementem terapii i znacznie wydłuża przeżycie wolne od choroby[12]. Przed wprowadzeniem chemioterapii samodzielne leczenie chirurgiczne, głównie za pomocą amputacji, prowadziło do 10–20% odsetku przeżyć pięcioletnich. Obecnie połączenie leczenia chirurgicznego i chemioterapii o intensywnym dawkowaniu prowadzi do osiągania przeżycia pięcioletniego u 60–70% leczonych[11].
Większość obecnych programów leczniczych obejmuje leczenie neoadiuwantowe, które ma za zadanie ułatwienie lub umożliwienie leczenia chirurgicznego[12], choć jednocześnie leczenie przedoperacyjne nie daje przewagi w wydłużeniu przeżycia całkowitego wobec samodzielnego leczenia adiuwantowego (pooperacyjnego)[12][150][151]. Wszyscy chorzy na kostniakomięsaka o wysokim stopniu złośliwości wymagają leczenia skojarzonego obejmującego neoadiuwantową (przedoperacyjną) chemioterapię (3–4 cykle), zabieg chirurgiczny i następnie adiuwantową (uzupełniającą) chemioterapię (3–6 cykli)[13][14].
W dwudziestopięcioletniej obserwacji u chorych na kostniakomięsaka o wysokiej złośliwości wykazano korzyść z leczenia okołooperacyjnego w wydłużeniu przeżycia całkowitego i przeżycia wolnego od choroby[152][153]. Jednak w przypadku kostniakomięsaka okostnowego nie ma dowodów wskazujących na korzyści w przeżyciu całkowitych chorych leczonych neoadiuwantowo[153][154][155].
Warunkiem kontynuacji przedoperacyjnej chemioterapii jest stwierdzenie co najmniej dobrej odpowiedzi histopatologicznej (≥90% martwicy utkania nowotworu), z kolei u chorych ze słabą i złą odpowiedzią (martwica <90%) należy rozważyć zmianę leczenia[153]. Jednak prawdopodobnie próby zmiany chemioterapii nie prowadzą do poprawy wyników końcowych leczenia[153][156][157][158][159].
W leczeniu I rzutu w leczeniu okołooperacyjnym oraz choroby przerzutowej zastosowanie znajdują[160]:
- cisplatyna z doksorubicyną,
- wysoka dawka metotreksatu, cisplatyna z doksorubicyną (program MAP),
- wysoka dawka metotreksatu, cisplatyna, doksorubicyna z ifosfamidem,
- cisplatyna, epirubicyna z ifosfamidem.
Muramylotripeptyd jest lekiem immunomodulującym. W jednym dużym randomizowanym badaniu wykazano, że u chorych poniżej 30. roku życia po radykalnej resekcji kostniakomięsaka lek dodany do wielolekowej chemioterapii zwiększa odsetek przeżyć sześcioletnich z 70% do 78%[12][161][162][163]. Obecnie, w związku z brakiem innych badań rola muramylotripeptydu w leczeniu kostniakomięsaka jest nieustalona[12][163]. Nie wykazano, aby dodanie IFN-α-2b do programu MAP poprawiało wyniki końcowe leczenia[164][165].
W przypadku nieuzyskania ujemnego marginesu resekcji może być wykonana reoperacja. Po reoperacji może być zastosowana radioterapia, która może poprawiać lokalną kontrolę choroby i wydłużać przeżycie całkowite[153][166]. Część chorych w starszym wieku może odnieść korzyści z leczenia chirurgicznego bez poprzedzającej chemioterapii[153].
W przypadku stwierdzenia nieoperacyjnego guza pomimo zastosowania neoadiuwantowej (przedoperacyjnej) chemioterapii zaleca się zastosowanie radioterapii lub adiuwantowej chemioterapii[165].
Leczenie choroby z przerzutami i zaawansowanej miejscowo
U około 10–20% chorych kostniakomięsak jest rozpoznawany w stadium choroby przerzutowej. Aktywność w kostniakomięsaku wykazują doksorubicyna, cisplatyna, cyklofosfamid, metotreksat w wysokich dawkach, ifosfamid, bleomycyna i daktynomycyna[167]. Obecne programy lecznicze są oparte o doksorubicynę, cisplatynę i metotreksat w wysokich dawkach[7][12][168].
W leczeniu I rzutu w leczeniu choroby przerzutowej oraz w leczeniu okołooperacyjnym jest stosowana[160]:
- cisplatyna z doksorubicyną,
- wysoka dawka metotreksatu z cisplatyną i doksorubicyną (program MAP),
- wysoka dawka metotreksatu z cisplatyną, doksorubicyną i ifosfamidem,
- cisplatyna z epirubicyną i ifosfamidem.
Badania kliniczne wykazały, że programy chemioterapii o intensywnej dawce oparte na doksorubicynie i cisplatynie z lub bez metotreksatu w wysokich dawkach dają porównywalne przeżycia długoterminowe jak programy wielolekowe[161][167][169][170][171][172][173]. Wykazano, że program dwulekowy zawierający cisplatynę i doksorubicynę jest lepiej tolerowany niż bardzo skomplikowany wielolekowy program T10 i nie wykazano różnicy w przeżyciu całkowitym[170]. W innym badaniu nie wykazano różnicy sześcioletniego przeżycia całkowitego pomiędzy programem trójlekowym zawierającym cisplatynę, doksorubicynę i metotreksat z programem czterolekowym zawierającym cisplatynę, doksorubicynę, metotreksat i ifosfamid[161].
W badaniu klinicznym porównano skuteczność połączenia doksorubicyny, metotreksatu i cisplatyny z połączeniem doksorubicyny, metotreksatu, daktynomycyny, cyklofosfamidu i bleomycyny, wykazano, że oba programy wywołują podobne przeżycie wolne od choroby (DFS), co przemawia za koniecznością dołączenia cisplatyny do chemioterapii kostniakomięsaka[174][175]. W innym badaniu stwierdzono, że odpowiedź histologiczna i czas wolny od obecności przerzutów był gorszy, gdy leczeni nie otrzymali doksorubicyny[175][176]. Skuteczność leczenia metotreksatem u dorosłych pozostaje przedmiotem sporów. W badaniu klinicznym porównano skuteczność połączenia doksorubicyny i cisplatyny z połączeniem doksorubicyny, cisplatyny i metotreksatu. Czas przeżycia całkowitego nie różnił się pomiędzy grupami, ale czas przeżycia wolny od choroby był lepszy w grupie chorych nieotrzymujących metotreksatu[169]. Jednak chorzy otrzymujący metotreksat otrzymali niższe dawki doksorubicyny i cisplatyny, co mogło przekładać się na wyniki końcowe[177]. U dorosłych trudniej niż u dzieci zastosować metotreksat w wysokich dawkach ze względu na towarzyszącą lekowi nefrotoksyczność[177]. Ifosfamid wykazuje znaczną aktywność przeciwnowotworową w leczeniu kostniakomięsaka. Jednak stwierdzono, że dodanie ifosfamidu do cisplatyny, doksorubicyny i metotreksatu nie zwiększa odsetka odpowiedzi histologicznych i przeżycia wolnego od zdarzeń (EFS)[178]. Również w innym badaniach dodanie ifosfamidu do doksorubicyny i cisplatyny nie zwiększyło przeżycia całkowitego leczonych[179][180].
U części chorych obecność przerzutów odległych nie wyklucza możliwości wyleczenia choroby[181]. W przypadku obecności pojedynczych, potencjalnie operacyjnych przerzutów do płuc, narządów miąższowych lub kości zaleca się przeprowadzenie przedoperacyjnej chemioterapii, a następnie wykonania metastazektomii. W przypadku nieoperacyjnych przerzutów konieczne jest leczenie ogólnoustrojowe, a następnie przeprowadza się powtórną ocenę operacyjności i ewentualnie wykonuje się zabieg[165]. Po operacji konieczna jest kontynuacja leczenia okołooperacyjnego[181].
W przypadku niektórych lokalizacji, szczególnie miedniczej i kręgowej, operacja nie jest możliwa i w leczeniu stosuje się chemioterapię i radioterapię. W wybranych przypadkach stosuje się chemioterapię dotętniczą, która może umożliwić chirurgiczną lokalną kontrolę choroby[181].
Leczenie choroby nawrotowej
U 30% chorych z chorobą zlokalizowaną i u 80% chorych z chorobą z przerzutami dochodzi do nawrotu choroby[182]. Nie wypracowano optymalnej strategii leczenia choroby nawrotowej, stosuje się programy lecznicze drugiego rzutu z lub bez reoperacji[183].
Do programów leczniczych stosowanych w chemioterapii II rzutu kostniakomięsaka zalicza się[160]:
- docetaksel z gemcytabiną,
- cyklofosfamid z etopozydem,
- ifosfamid z lub bez etopozydem,
- cyklofosfamid z topotekanem,
- gemcytabinę w monoterapii,
- metotreksat, etopozyd i ifosfamid,
- 153Sm-EDTMP,
- Ra223,
- sorafenib,
- sorafenib z ewerolimusem.
Połączenie etopozydu z cyklofosfamidem lub ifosfamidem oceniono w kilku badaniach klinicznych. We francuskim badaniu połączenie etopozydu z ifosfamidem wywoływało odpowiedź u 48% leczonych[184]. W innym badaniu połączenie etopozydu z cyklofosfamidem wywoływało odpowiedź u 19% leczonych i prowadziło do stabilizacji choroby u 35% leczonych[185]. Gemcytabina w monoterapii[186], połączenie gemcytabiny z docetakselem[187], połączenie ifosfamidu, karboplatyny i etopozydu[188], połączenie cyklofosfamidu z topotekanem[189] wykazują aktywność w leczeniu kostniakomięsaka.
153Sm-EDTMP jest radiofarmaceutykiem emitującym promieniowanie beta stosowanym w leczeniu przerzutów do kości[190][191]. Badania sugerują, że Ra223 emitujący promieniowanie alfa może być skuteczny w leczeniu kostniakomięsaka i wykazywać niższą mielotoksyczność[192][182].
Sorafenib jest inhibitorem kilku kinaz. W badaniu klinicznym oceniono skuteczność leku i stwierdzono 4-miesięczną medianę przeżycia wolnego od progresji (PFS)[193]. W innym badaniu wykazano aktywność sorafenibu połączeniu z ewerolimusem, jednak połączenie wykazuje znaczną toksyczność[194].
Rokowanie
Szacuje się, że odsetek pięcioletniego przeżycia chorych na kostniakomięsaka wynosi około 65–68%[1][11][195]. Odsetek przeżycia całkowitego zależy od stadium choroby, lokalizacji guza, płci i od wieku zachorowania[78]. Obecność przerzutów znacznie pogarsza rokowanie. Chorzy z chorobą zlokalizowaną osiągają 75% odsetek pięcioletniego przeżycia, podczas gdy odsetek przeżyć pięcioletnich u chorych z obecnymi przerzutami odległymi spada do 21%[195]. Nawrót choroby jest obserwowany u 30–40% chorych z pierwotnie zlokalizowaną chorobą[40][195], z czego w 80% przypadków występuje jako pojawienie się przerzutów do płuc[195]. Rokowanie jest lepsze w lokalizacji kostniakomięsaka w obrębie kości długich, z kolei lokalizacja w obrębie miednicy czy kręgosłupa wiąże się ze znacznie gorszym rokowaniem[78][3][196]. Niektóre podtypy histopatologiczne różnią się rokowaniem, jednak ze względu na ich stosunkową rzadkość i niewielkie przebadane grupy chorych faktyczna ocena jest trudna. Kostniakomięsak przykostny wiąże się z lepszym rokowaniem, a z kolei kostniakomięsak związany z chorobą Pageta ze znacznie gorszym[78]. Obserwuje się spadek odsetku pięcioletniego przeżycia po 50. roku życia[78][3][197][198][199][200]. Kobiety osiągają lepsze przeżycie niż mężczyźni[78].
Kostniakomięsak u zwierząt
Kostniakomięsak jest nowotworem złośliwym rozpoznanym u wielu gatunków zwierząt: niektórych gadów[201], niektórych ptaków[202][203][204][205][206], niektórych ssaków, w tym gryzonii[207][208][209][210][211][212], zającowatych[213][214], łasicowatych[215], jeżowatych[216], wołowatych[217][218], świniowatych[219], wielbłądowatych[220], koniowatych[221][222][223], niedźwiedziowatych[224] i niektórych naczelnych[225][226].
Etiologia kostniakomięsaka u zwierząt nie jest znana, zidentyfikowano kilka czynników wpływających na patogenezę choroby[227]. Teoria wirusowa nie została potwierdzona, choć występowanie kostniakomięsaka u psów z tego samego miotu oraz możliwość wywołania choroby poprzez sztuczne wprowadzenie komórek nowotworowych sugerowały taką możliwość. Niektórzy autorzy sugerują udział powtarzalnych urazów okolicy przynasadowej. Opisano przypadki kostniakomięsaka u zwierząt poddanych radioterapii[228]. Histologicznie kostniakomięsak jest klasyfikowany w oparciu o klasyfikację WHO stosowaną u ludzi[227]. Wyróżnia się podtyp osteoblastyczny, fibroblastyczny, chondroblastyczny, naczyniakowaty, olbrzymiokomórkowy i niskozróżnicowany[228][229].
Głównym objawem jest bolesność zajętej okolicy, obrzęk, deformacja i zaburzenie funkcji zajętej struktury. Mogą być obecne zaniki mięśniowe oraz powiększone okoliczne węzły chłonne. Rzadko pierwszym objawem jest złamanie patologiczne. W obrębie żuchwy i szczęki nowotwór może powodować zaburzenie połykania, ślinotok, utratę chęci do jedzenia. Guzy zatok i jamy nosowej są przyczyną patologicznego wycieku z jamy nosowej i zaburzeń oddechowych[228][230]. Rozpoznanie jest stawiane na podstawie badania klinicznego, zdjęcia RTG w dwóch projekcjach i badania histopatologicznego[226][228]. W RTG w obrębie guza mogą być widoczne zmiany wytwórcze, jak i osteolityczne. Zdjęcie RTG płuc pozwala na ocenę obecności przerzutów. Do rozpoznania histopatologicznego konieczne jest uzyskanie reprezentatywnego materiału z guza drogą biopsji gruboigłowej lub preparatu pooperacyjnego[228][231].
Podstawową metodą leczenia jest postępowanie chirurgiczne. Ma ono na celu lokalną kontrolę choroby i zmniejszenie dolegliwości bólowych u zwierzęcia[232]. W kostniakomięsaku kończyn zwykle wykonuje się amputacje, rzadziej wykonuje się zabiegi oszczędzające kończynę[226][228]. Amputacja jest zabiegiem krótkim, niesie ze sobą niższe ryzyko powikłań i jest względnie niskokosztową procedurą. Nie wykonuje się amputacji w przypadku obecności masywnych przerzutów do płuc. Operacje zachowujące kończynę bywają stosowane głównie w guzach zlokalizowanych w dystalnej części kości promieniowej. Również w zajęciu szkieletu osiowego główną metodą leczenia jest operacja chirurgiczna o charakterze zależnym od konkretnej lokalizacji[228]. U psów chemioterapia bywa stosowana w leczeniu uzupełniającym po zabiegu chirurgicznym lub w leczeniu paliatywnym[228][233]. W chemioterapii zastosowanie znajduje cisplatyna lub karboplatyna albo doksorubicyna[228][234]. U kotów nie stosuje się cisplatyny[233]. Leki mogą być podawane w monoterapii, rzadziej w politerapii[234]. W przypadkach zaawansowanej choroby może być stosowana chemioterapia paliatywna – stosuje się cisplatynę, doksorubicynę lub mitoksantron w monoterapii[228]. W paliatywnym leczeniu bólu bywa stosowana radioterapia[235].
Przypisy
- ↑ a b c d e G. Ottaviani, N. Jaffe. The epidemiology of osteosarcoma. „Cancer Treat Res”. 152, s. 3–13, 2009. DOI: 10.1007/978-1-4419-0284-9_1. PMID: 20213383.
- ↑ Vinay Kumar, Ramzi S. Cotran, Stanley L. Robins: Robins Patologia. Wrocław: Elsevier Urban & Partner, 2005, s. 875–876. ISBN 83-89581-92-2.
- ↑ a b c d L. Mirabello, R.J. Troisi, S.A. Savage. Osteosarcoma incidence and survival rates from 1973 to 2004: data from the Surveillance, Epidemiology, and End Results Program. „Cancer”. 115 (7), s. 1531–1543, Apr 2009. DOI: 10.1002/cncr.24121. PMID: 19197972.
- ↑ a b c d e P. Picci. Osteosarcoma (osteogenic sarcoma). „Orphanet J Rare Dis”. 2, s. 6, 2007. DOI: 10.1186/1750-1172-2-6. PMID: 17244349.
- ↑ R.S. Brodey. The use of naturally occurring cancer in domestic animals for research into human cancer: general considerations and a review of canine skeletal osteosarcoma. „Yale J Biol Med”. 52 (4). s. 345–361. PMID: 115162.
- ↑ a b E. Harris Randall: Global Epidemiology of Cancer. Jones & Bartlett Publishers, 2015. ISBN 978-1-284-03446-2.
- ↑ a b c d e f g h i j k l m n o p q r s t u v w x D.S. Geller, R. Gorlick. Osteosarcoma: a review of diagnosis, management, and treatment strategies. „Clin Adv Hematol Oncol”. 8 (10), s. 705–718, Oct 2010. PMID: 21317869.
- ↑ a b c d e Andrew L. Folpe, Carrie Y. Inwards: Bone and Soft Tissue Pathology: A Volume in the Foundations in Diagnostic Pathology Series. Elsevier Health Sciences, 2009, s. 321. ISBN 978-1-4377-1947-5.
- ↑ a b c d e f Casali i in. 2014 ↓, s. 1.
- ↑ a b c d Rutkowski i Warzocha 2013 ↓, s. 443.
- ↑ a b c X. Dai, W. Ma, X. He, R.K. Jha. Review of therapeutic strategies for osteosarcoma, chondrosarcoma, and Ewing’s sarcoma. „Med Sci Monit”. 17 (8), s. RA177-190, Aug 2011. PMID: 21804475.
- ↑ a b c d e f g h i Casali i in. 2014 ↓, s. 3.
- ↑ a b c Krzakowski i in. 2015 ↓, s. 923.
- ↑ a b c Rutkowski i Warzocha 2013 ↓, s. 450.
- ↑ a b Rutkowski i Warzocha 2013 ↓, s. 441–442.
- ↑ M.P. Mohanta. Growing pains: practitioners’ dilemma. „Indian Pediatr”. 51 (5), s. 379–383, May 2014. PMID: 24953579.
- ↑ mp: Ból wzrostowy u dzieci. [dostęp 2016-01-31].
- ↑ Y. Uziel, P.J. Hashkes. Growing pains in children. „Pediatr Rheumatol Online J”. 5, s. 5, 2007. DOI: 10.1186/1546-0096-5-5. PMID: 17550631.
- ↑ a b c d Fletcher, Unni i Mertens 2002 ↓, s. 265.
- ↑ a b c Philip Lanzkowsky: Manual of Pediatric Hematology and Oncology. Academic Press, 2005, s. 742. ISBN 978-0-12-375155-3.
- ↑ Fletcher, Unni i Mertens 2002 ↓, s. 264–265.
- ↑ Timothy A. Damron: Oncology and Basic Science. Lippincott Williams & Wilkins, 2008, s. 181. ISBN 978-0-7817-8045-2.
- ↑ a b c d e f L. Mirabello, R.J. Troisi, S.A. Savage. International osteosarcoma incidence patterns in children and adolescents, middle ages and elderly persons. „Int J Cancer”. 125 (1), s. 229–234, Jul 2009. DOI: 10.1002/ijc.24320. PMID: 19330840.
- ↑ a b Stuart H. Orkin, David G. Natha, David Ginsburg, A. Thomas Look, David E. Fisher, Samuel Lux IV: Nathan and Oski’s Hematology and Oncology of Infancy and Childhoo. Elsevier Health Sciences, 2014, s. 2019. ISBN 978-0-323-29177-4.
- ↑ Daniela Stefan, Carlos Rodriguez-Galindo: Pediatric Hematology-Oncology in Countries with Limited Resources: A Practical Manual. Springer Science & Business Media, 2013, s. 323. ISBN 978-1-4614-3891-5.
- ↑ Rutkowski i Warzocha 2013 ↓, s. 440.
- ↑ Fletcher, Unni i Mertens 2002 ↓, s. 226.
- ↑ a b c d e f g h i j k Z.S. Kundu. Classification, imaging, biopsy and staging of osteosarcoma. „Indian J Orthop”. 48 (3), s. 238–246, May 2014. DOI: 10.4103/0019-5413.132491. PMID: 24932027.
- ↑ a b Krzakowski i in. 2015 ↓, s. 883.
- ↑ a b Alberto S. Pappo: Pediatric Bone and Soft Tissue Sarcomas. Springer Science & Business Media, 2006, s. 222. ISBN 978-3-540-29447-4.
- ↑ Czerniak 2015 ↓, s. 262–271.
- ↑ John D. Reith: Current Concepts in Bone Pathology, an Issue of Surgical Pathology Clinics. Elsevier Health Sciences, 2012, s. 301.
- ↑ a b c d Robert F. Todd, Kathleen A. Cooney, Teresa G. Hayes, Martha Pritchett Mims, Francis P. Worden: Tumor Board Review, Second Edition: Guideline and Case Reviews in Oncology. Demos Medical Publishing, 2015, s. 254. ISBN 978-1-62070-060-0.
- ↑ R. Elhasid, E. Vlodavsky, A. Nachtigal, Z. Keidar i inni. Case 3. Bone marrow involvement in osteosarcoma. „J Clin Oncol”. 19 (1), s. 276–278, Jan 2001. PMID: 11134225.
- ↑ a b DeVita, Lawrence i Rosenberg 2008 ↓, s. 1797.
- ↑ a b c V.A. Currall, J.H. Dixon. Synchronous multifocal osteosarcoma: case report and literature review. „Sarcoma”. 2006, s. 53901, 2006. DOI: 10.1155/SRCM/2006/53901. PMID: 17251658.
- ↑ Adam Greenspan, Gernot Jundt, Wolfgang Remagen: Differential Diagnosis in Orthopaedic Oncology. Lippincott Williams & Wilkins, 2007, s. 108. ISBN 978-0-7817-7930-2.
- ↑ a b H. Cho, B.J. Park, Y.K. Park. Multifocal osteosarcoma of the skull: multiple primary or metastatic? A case report. „Korean J Pathol”. 48 (2), s. 146–150, Apr 2014. DOI: 10.4132/KoreanJPathol.2014.48.2.146. PMID: 24868228.
- ↑ DeVita, Lawrence i Rosenberg 2008 ↓, s. 1797–1798.
- ↑ a b c Paige Bennett, Umesh D. Oza: Diagnostic Imaging: Nuclear Medicine. Elsevier Health Sciences, 2015, s. 462. ISBN 978-0-323-40044-2.
- ↑ a b c Fletcher, Unni i Mertens 2002 ↓, s. 264.
- ↑ Stuart H. Orkin, David G. Nathan, David Ginsburg, A. Thomas Look, David E. Fisher, Samuel Lux IV, David E. Fisher: Oncology of Infancy and Childhood. Elsevier Health Sciences, 2009, s. 879. ISBN 978-1-4377-2138-6.
- ↑ Edward Estlin, Richard Gilbertson, Rob Wynn: Pediatric Hematology and Oncology: Scientific Principles and Clinical Practice. John Wiley & Sons, 2011. ISBN 978-1-4443-2255-2.
- ↑ a b c d e Fletcher 2007 ↓, s. 1616.
- ↑ a b Bui i Dodd 2014 ↓, s. 283.
- ↑ Fletcher 2007 ↓, s. 1617.
- ↑ Bui i Dodd 2014 ↓, s. 284.
- ↑ a b Fletcher, Unni i Mertens 2002 ↓, s. 266.
- ↑ Fletcher, Unni i Mertens 2002 ↓, s. 271.
- ↑ a b Fletcher 2007 ↓, s. 1619.
- ↑ Bui i Dodd 2014 ↓, s. 288.
- ↑ a b Fletcher, Unni i Mertens 2002 ↓, s. 271–272.
- ↑ Fletcher, Unni i Mertens 2002 ↓, s. 272.
- ↑ a b c d Fletcher, Unni i Mertens 2002 ↓, s. 273.
- ↑ Fletcher 2007 ↓, s. 1620.
- ↑ a b Fletcher, Unni i Mertens 2002 ↓, s. 275.
- ↑ Thomas Pope, Hans L. Bloem, Javier Beltran, William B. Morrison, David John Wilson: Musculoskeletal Imaging. Elsevier Health Sciences, 2014, s. 928. ISBN 978-0-323-27818-8.
- ↑ Fletcher, Unni i Mertens 2002 ↓, s. 276.
- ↑ a b c d e Fletcher, Unni i Mertens 2002 ↓, s. 279.
- ↑ a b c Unni i Inwards 2010 ↓, s. 165.
- ↑ Fletcher, Unni i Mertens 2002 ↓, s. 279–280.
- ↑ Fletcher, Unni i Mertens 2002 ↓, s. 280.
- ↑ Unni i Inwards 2010 ↓, s. 167.
- ↑ Fletcher, Unni i Mertens 2002 ↓, s. 282.
- ↑ Vincent J. Vigorita Vincent J. Vigorita: Orthopaedic Pathology. Lippincott Williams & Wilkins, 2007, s. 303. ISBN 978-0-7817-9670-5.
- ↑ a b c Bui i Dodd 2014 ↓, s. 297.
- ↑ a b c Fletcher, Unni i Mertens 2002 ↓, s. 283.
- ↑ Vincent J. Vigorita Vincent J. Vigorita: Orthopaedic Pathology. Lippincott Williams & Wilkins, 2007, s. 306. ISBN 978-0-7817-9670-5.
- ↑ a b c V.S. Kumar, N. Barwar, S.A. Khan. Surface osteosarcomas: Diagnosis, treatment and outcome. „Indian J Orthop”. 48 (3), s. 255–261, May 2014. DOI: 10.4103/0019-5413.132503. PMID: 24932030.
- ↑ a b Fletcher, Unni i Mertens 2002 ↓, s. 284.
- ↑ a b c d e Fletcher, Unni i Mertens 2002 ↓, s. 182.
- ↑ a b c M. Hoch, S. Ali, S. Agrawal, C. Wang i inni. Extraskeletal osteosarcoma: a case report and review of the literature. „J Radiol Case Rep”. 7 (7), s. 15–23, Jul 2013. DOI: 10.3941/jrcr.v7i7.1245. PMID: 24421944.
- ↑ Czerniak 2015 ↓, s. 345.
- ↑ a b Andrew L. Folpe, Carrie Y. Inwards: Bone and Soft Tissue Pathology. Elsevier Health Sciences, 2010, s. 243–245. ISBN 978-0-443-06688-7.
- ↑ a b c d e f g h i j k l J.W. Martin, J.A. Squire, M. Zielenska. The genetics of osteosarcoma. „Sarcoma”. 2012, s. 627254, 2012. DOI: 10.1155/2012/627254. PMID: 22685381.
- ↑ a b c Jaffe, Bruland i Bielack 2010 ↓, s. 24.
- ↑ a b R. Gorlick, C. Khanna. Osteosarcoma. „J Bone Miner Res”. 25 (4), s. 683–691, Apr 2010. DOI: 10.1002/jbmr.77. PMID: 20205169.
- ↑ a b c d e f S.A. Savage, L. Mirabello. Using epidemiology and genomics to understand osteosarcoma etiology. „Sarcoma”. 2011, s. 548151, 2011. DOI: 10.1155/2011/548151. PMID: 21437228.
- ↑ Jaffe, Bruland i Bielack 2010 ↓, s. 19–20.
- ↑ S.J. Cotterill, C.M. Wright, M.S. Pearce, A.W. Craft. Stature of young people with malignant bone tumors. „Pediatr Blood Cancer”. 42 (1), s. 59–63, Jan 2004. DOI: 10.1002/pbc.10437. PMID: 14752796.
- ↑ Jaffe, Bruland i Bielack 2010 ↓, s. 20.
- ↑ Jaffe, Bruland i Bielack 2010 ↓, s. 19.
- ↑ R.A. Tjalma. Canine bone sarcoma: estimation of relative risk as a function of body size. „J Natl Cancer Inst”. 36 (6), s. 1137–1150, Jun 1966. PMID: 5221997.
- ↑ M.F. Hansen, M. Seton, A. Merchant. Osteosarcoma in Paget’s disease of bone. „J Bone Miner Res”. 21 Suppl 2, s. P58-63, Dec 2006. DOI: 10.1359/jbmr.06s211. PMID: 17229010.
- ↑ a b Jaffe, Bruland i Bielack 2010 ↓, s. 25.
- ↑ Terrance D. Peabody, Samer Attar: Orthopaedic Oncology: Primary and Metastatic Tumors of the Skeletal System. Springer, 2014, s. 69. ISBN 978-3-319-07323-1.
- ↑ A. Longhi, S. Ferrari, A. Tamburini, R. Luksch i inni. Late effects of chemotherapy and radiotherapy in osteosarcoma and Ewing sarcoma patients: the Italian Sarcoma Group Experience (1983–2006). „Cancer”. 118 (20), s. 5050–5059, Oct 2012. DOI: 10.1002/cncr.27493. PMID: 22415578.
- ↑ Jaffe, Bruland i Bielack 2010 ↓, s. 26.
- ↑ T.O. Henderson, J. Whitton, M. Stovall, A.C. Mertens i inni. Secondary sarcomas in childhood cancer survivors: a report from the Childhood Cancer Survivor Study. „J Natl Cancer Inst”. 99 (4), s. 300–308, Feb 2007. DOI: 10.1093/jnci/djk052. PMID: 17312307.
- ↑ Jaffe, Bruland i Bielack 2010 ↓, s. 28.
- ↑ Jaffe, Bruland i Bielack 2010 ↓, s. 21–22.
- ↑ a b P. McQueen, S. Ghaffar, Y. Guo, E.M. Rubin i inni. The Wnt signaling pathway: implications for therapy in osteosarcoma. „Expert Rev Anticancer Ther”. 11 (8), s. 1223–1232, Aug 2011. DOI: 10.1586/era.11.94. PMID: 21916576.
- ↑ J. Tian, H. He, G. Lei. Wnt/β-catenin pathway in bone cancers. „Tumour Biol”. 35 (10), s. 9439–9445, Oct 2014. DOI: 10.1007/s13277-014-2433-8. PMID: 25117074.
- ↑ R.C. Haydon, A. Deyrup, A. Ishikawa, R. Heck i inni. Cytoplasmic and/or nuclear accumulation of the beta-catenin protein is a frequent event in human osteosarcoma. „Int J Cancer”. 102 (4), s. 338–342, Dec 2002. DOI: 10.1002/ijc.10719. PMID: 12402302.
- ↑ a b c d e J.P. He, Y. Hao, X.L. Wang, X.J. Yang i inni. Review of the molecular pathogenesis of osteosarcoma. „Asian Pac J Cancer Prev”. 15 (15), s. 5967–5976, 2014. PMID: 25124559.
- ↑ F.X. Dieudonné, A. Marion, P.J. Marie, D. Modrowski. Targeted inhibition of T-cell factor activity promotes syndecan-2 expression and sensitization to doxorubicin in osteosarcoma cells and bone tumors in mice. „J Bone Miner Res”. 27 (10), s. 2118–2129, Oct 2012. DOI: 10.1002/jbmr.1650. PMID: 22550000.
- ↑ F.X. Dieudonné, A. Marion, E. Haÿ, P.J. Marie i inni. High Wnt signaling represses the proapoptotic proteoglycan syndecan-2 in osteosarcoma cells. „Cancer Res”. 70 (13), s. 5399–5408, Jul 2010. DOI: 10.1158/0008-5472.CAN-10-0090. PMID: 20530678.
- ↑ B.P. Himelstein, N. Asada, M.R. Carlton, M.H. Collins. Matrix metalloproteinase-9 (MMP-9) expression in childhood osseous osteosarcoma. „Med Pediatr Oncol”. 31 (6), s. 471–474, Dec 1998. PMID: 9835898.
- ↑ a b c d e J. Zhang, X.H. Yu, Y.G. Yan, C. Wang i inni. PI3K/Akt signaling in osteosarcoma. „Clin Chim Acta”. 444, s. 182–192, Apr 2015. DOI: 10.1016/j.cca.2014.12.041. PMID: 25704303.
- ↑ W. Liang, B. Gao, G. Xu, D. Weng i inni. Possible contribution of aminopeptidase N (APN/CD13) to migration and invasion of human osteosarcoma cell lines. „Int J Oncol”. 45 (6), s. 2475–2485, Dec 2014. DOI: 10.3892/ijo.2014.2664. PMID: 25340499.
- ↑ a b c M.L. Broadhead, J.C. Clark, D.E. Myers, C.R. Dass i inni. The molecular pathogenesis of osteosarcoma: a review. „Sarcoma”. 2011, s. 959248, 2011. DOI: 10.1155/2011/959248. PMID: 21559216.
- ↑ W. Fu, L. Ma, B. Chu, X. Wang i inni. The cyclin-dependent kinase inhibitor SCH 727965 (dinacliclib) induces the apoptosis of osteosarcoma cells. „Mol Cancer Ther”. 10 (6), s. 1018–1027, Jun 2011. DOI: 10.1158/1535-7163.MCT-11-0167. PMID: 21490307.
- ↑ A.B. Mohseny, K. Szuhai, S. Romeo, E.P. Buddingh i inni. Osteosarcoma originates from mesenchymal stem cells in consequence of aneuploidization and genomic loss of Cdkn2. „J Pathol”. 219 (3), s. 294–305, Nov 2009. DOI: 10.1002/path.2603. PMID: 19718709.
- ↑ C. Stock, L. Kager, F.M. Fink, H. Gadner i inni. Chromosomal regions involved in the pathogenesis of osteosarcomas. „Genes Chromosomes Cancer”. 28 (3), s. 329–336, Jul 2000. PMID: 10862039.
- ↑ G. Han, Y. Wang, W. Bi. C-Myc overexpression promotes osteosarcoma cell invasion via activation of MEK-ERK pathway. „Oncol Res”. 20 (4), s. 149–156, 2012. PMID: 23461061.
- ↑ A. Franchi, L. Arganini, G. Baroni, A. Calzolari i inni. Expression of transforming growth factor beta isoforms in osteosarcoma variants: association of TGF beta 1 with high-grade osteosarcomas. „J Pathol”. 185 (3), s. 284–289, Jul 1998. DOI: <284::AID-PATH94>3.0.CO;2-Z 10.1002/(SICI)1096-9896(199807)185:3<284::AID-PATH94>3.0.CO;2-Z. PMID: 9771482.
- ↑ D. Nikitovic, A. Zafiropoulos, P. Katonis, A. Tsatsakis i inni. Transforming growth factor-beta as a key molecule triggering the expression of versican isoforms v0 and v1, hyaluronan synthase-2 and synthesis of hyaluronan in malignant osteosarcoma cells. „IUBMB Life”. 58 (1), s. 47–53, Jan 2006. DOI: 10.1080/15216540500531713. PMID: 16540432.
- ↑ F. Li, S. Li, T. Cheng. TGF-β1 promotes osteosarcoma cell migration and invasion through the miR-143-versican pathway. „Cell Physiol Biochem”. 34 (6), s. 2169–2179, 2014. DOI: 10.1159/000369660. PMID: 25562163.
- ↑ H. Nishimura, O. Nakamura, Y. Yamagami, M. Mori i inni. GSK-3 inhibitor inhibits cell proliferation and induces apoptosis in human osteosarcoma cells. „Oncol Rep”. 35 (4), s. 2348–2354, Apr 2016. DOI: 10.3892/or.2016.4565. PMID: 26781995.
- ↑ Q.L. Tang, X.B. Xie, J. Wang, Q. Chen i inni. Glycogen synthase kinase-3β, NF-κB signaling, and tumorigenesis of human osteosarcoma. „J Natl Cancer Inst”. 104 (10), s. 749–763, May 2012. DOI: 10.1093/jnci/djs210. PMID: 22534782.
- ↑ K. Nishijo, T. Nakayama, T. Aoyama, T. Okamoto i inni. Mutation analysis of the RECQL4 gene in sporadic osteosarcomas. „Int J Cancer”. 111 (3), s. 367–372, Sep 2004. DOI: 10.1002/ijc.20269. PMID: 15221963.
- ↑ A.J. Ng, M.K. Walia, M.F. Smeets, A.J. Mutsaers i inni. The DNA helicase recql4 is required for normal osteoblast expansion and osteosarcoma formation. „PLoS Genet”. 11 (4), s. e1005160, Apr 2015. DOI: 10.1371/journal.pgen.1005160. PMID: 25859855.
- ↑ H.A. Siitonen, J. Sotkasiira, M. Biervliet, A. Benmansour i inni. The mutation spectrum in RECQL4 diseases. „Eur J Hum Genet”. 17 (2), s. 151–158, Feb 2009. DOI: 10.1038/ejhg.2008.154. PMID: 18716613.
- ↑ J. German. Bloom’s syndrome. XX. The first 100 cancers. „Cancer Genet Cytogenet”. 93 (1), s. 100–106, Jan 1997. PMID: 9062585.
- ↑ M. Goto, R.W. Miller, Y. Ishikawa, H. Sugano. Excess of rare cancers in Werner syndrome (adult progeria). „Cancer Epidemiol Biomarkers Prev”. 5 (4), s. 239–246, Apr 1996. PMID: 8722214.
- ↑ Q.X. Paulson, R.V. Pusapati, S. Hong, R.L. Weaks i inni. Transgenic expression of E2F3a causes DNA damage leading to ATM-dependent apoptosis. „Oncogene”. 27 (36), s. 4954–4961, Aug 2008. DOI: 10.1038/onc.2008.138. PMID: 18469863.
- ↑ C.D. Hurst, D.C. Tomlinson, S.V. Williams, F.M. Platt i inni. Inactivation of the Rb pathway and overexpression of both isoforms of E2F3 are obligate events in bladder tumours with 6p22 amplification. „Oncogene”. 27 (19), s. 2716–2727, Apr 2008. DOI: 10.1038/sj.onc.1210934. PMID: 18037967.
- ↑ Y. Liao, Y. Feng, J. Shen, Y. Gao i inni. Clinical and biological significance of PIM1 kinase in osteosarcoma. „J Orthop Res”, Dec 2015. DOI: 10.1002/jor.23134. PMID: 26687194.
- ↑ J. Yang, D. Yang, Y. Sun, B. Sun i inni. Genetic amplification of the vascular endothelial growth factor (VEGF) pathway genes, including VEGFA, in human osteosarcoma. „Cancer”. 117 (21), s. 4925–4938, Nov 2011. DOI: 10.1002/cncr.26116. PMID: 21495021.
- ↑ M.M. Bui, T.K. Bagui, D.C. Boulware, D.G. Letson i inni. Altered expression of cell cycle regulatory proteins in benign and malignant bone and soft tissue neoplasms. „In Vivo”. 21 (5). s. 729–737. PMID: 18019405.
- ↑ X.Y. Lu, Y. Lu, Y.J. Zhao, K. Jaeweon i inni. Cell cycle regulator gene CDC5L, a potential target for 6p12-p21 amplicon in osteosarcoma. „Mol Cancer Res”. 6 (6), s. 937–946, Jun 2008. DOI: 10.1158/1541-7786.MCR-07-2115. PMID: 18567798.
- ↑ J.B. Lian, A. Javed, S.K. Zaidi, C. Lengner i inni. Regulatory controls for osteoblast growth and differentiation: role of Runx/Cbfa/AML factors. „Crit Rev Eukaryot Gene Expr”. 14 (1–2), s. 1–41, 2004. PMID: 15104525.
- ↑ S. Mendoza, H. David, G.M. Gaylord, C.W. Miller. Allelic loss at 10q26 in osteosarcoma in the region of the BUB3 and FGFR2 genes. „Cancer Genet Cytogenet”. 158 (2), s. 142–147, Apr 2005. DOI: 10.1016/j.cancergencyto.2004.08.035. PMID: 15796961.
- ↑ a b c d e f Krzakowski i in. 2015 ↓, s. 913.
- ↑ a b Krzakowski i in. 2015 ↓, s. 911.
- ↑ a b c Bogdan Pruszyński, Bogusława Benendo-Kapuścińska: Radiologia: diagnostyka obrazowa: Rtg, TK, USG, MR i radioizotopy. PZWL, 2008, s. 407. ISBN 978-83-200-3666-4.
- ↑ A.M. Aisen, W. Martel, E.M. Braunstein, K.I. McMillin i inni. MRI and CT evaluation of primary bone and soft-tissue tumors. „AJR Am J Roentgenol”. 146 (4), s. 749–756, Apr 1986. DOI: 10.2214/ajr.146.4.749. PMID: 3485348.
- ↑ K. Bohndorf, M. Reiser, B. Lochner, W. Féaux de Lacroix i inni. Magnetic resonance imaging of primary tumours and tumour-like lesions of bone. „Skeletal Radiol”. 15 (7), s. 511–517, 1986. PMID: 3775414.
- ↑ Jaffe, Bruland i Bielack 2010 ↓, s. 73.
- ↑ Casali i in. 2014 ↓, s. 2.
- ↑ Rutkowski i Warzocha 2013 ↓, s. 446.
- ↑ Krzakowski i in. 2015 ↓, s. 916.
- ↑ Rutkowski i Warzocha 2013 ↓, s. 447.
- ↑ Krzakowski i in. 2015 ↓, s. 917.
- ↑ a b c d e G. Bacci, S. Ferrari, M. Mercuri, F. Bertoni i inni. Predictive factors for local recurrence in osteosarcoma: 540 patients with extremity tumors followed for minimum 2.5 years after neoadjuvant chemotherapy. „Acta Orthop Scand”. 69 (3), s. 230–236, Jun 1998. PMID: 9703394.
- ↑ a b c d DeVita, Lawrence i Rosenberg 2008 ↓, s. 1802.
- ↑ a b c d e DeVita, Lawrence i Rosenberg 2008 ↓, s. 1803.
- ↑ a b Rutkowski i Warzocha 2013 ↓, s. 448.
- ↑ a b Krzakowski i in. 2015 ↓, s. 919.
- ↑ DeVita, Lawrence i Rosenberg 2008 ↓, s. 1816–1819.
- ↑ DeVita, Lawrence i Rosenberg 2008 ↓, s. 1820.
- ↑ B.T. Rougraff, M.A. Simon, J.S. Kneisl, D.B. Greenberg i inni. Limb salvage compared with amputation for osteosarcoma of the distal end of the femur. A long-term oncological, functional, and quality-of-life study. „J Bone Joint Surg Am”. 76 (5), s. 649–656, May 1994. PMID: 8175811.
- ↑ a b DeVita, Lawrence i Rosenberg 2008 ↓, s. 1804.
- ↑ a b DeVita, Lawrence i Rosenberg 2008 ↓, s. 1806.
- ↑ a b L.M. Nystrom, J.A. Morcuende. Expanding endoprosthesis for pediatric musculoskeletal malignancy: current concepts and results. „Iowa Orthop J”. 30, s. 141–149, 2010. PMID: 21045986.
- ↑ Mikel San-Julian: Cañadell’s Pediatric Bone Sarcomas: Epiphysiolysis before Excision. Springer, 2016, s. 92. ISBN 978-3-319-24220-0.
- ↑ R. Kotz. Rotationplasty. „Semin Surg Oncol”. 13 (1). s. 34–40. PMID: 9025180.
- ↑ a b c B. Fuchs, F.H. Sim. Rotationplasty about the knee: surgical technique and anatomical considerations. „Clin Anat”. 17 (4), s. 345–353, May 2004. DOI: 10.1002/ca.10211. PMID: 15108342.
- ↑ Krzakowski i in. 2015 ↓, s. 925.
- ↑ DeVita, Lawrence i Rosenberg 2008 ↓, s. 1808.
- ↑ A.M. Goorin, D.J. Schwartzentruber, M. Devidas, M.C. Gebhardt i inni. Presurgical chemotherapy compared with immediate surgery and adjuvant chemotherapy for nonmetastatic osteosarcoma: Pediatric Oncology Group Study POG-8651. „J Clin Oncol”. 21 (8), s. 1574–1580, Apr 2003. DOI: 10.1200/JCO.2003.08.165. PMID: 12697883.
- ↑ N.M. Bernthal, N. Federman, F.R. Eilber, S.D. Nelson i inni. Long-term results (>25 years) of a randomized, prospective clinical trial evaluating chemotherapy in patients with high-grade, operable osteosarcoma. „Cancer”. 118 (23), s. 5888–5893, Dec 2012. DOI: 10.1002/cncr.27651. PMID: 22648705.
- ↑ a b c d e f Biermann i in. 2016 ↓, s. 53.
- ↑ M. Cesari, M. Alberghini, D. Vanel, E. Palmerini i inni. Periosteal osteosarcoma: a single-institution experience. „Cancer”. 117 (8), s. 1731–1735, Apr 2011. DOI: 10.1002/cncr.25718. PMID: 21472720.
- ↑ R.J. Grimer, S. Bielack, S. Flege, S.R. Cannon i inni. Periosteal osteosarcoma-a European review of outcome. „Eur J Cancer”. 41 (18), s. 2806–2811, Dec 2005. DOI: 10.1016/j.ejca.2005.04.052. PMID: 16290134.
- ↑ S. Smeland, C. Müller, T.A. Alvegard, T. Wiklund i inni. Scandinavian Sarcoma Group Osteosarcoma Study SSG VIII: prognostic factors for outcome and the role of replacement salvage chemotherapy for poor histological responders. „Eur J Cancer”. 39 (4), s. 488–494, Mar 2003. PMID: 12751380.
- ↑ S. Smeland, O.S. Bruland, L. Hjorth, O. Brosjö i inni. Results of the Scandinavian Sarcoma Group XIV protocol for classical osteosarcoma: 63 patients with a minimum follow-up of 4 years. „Acta Orthop”. 82 (2), s. 211–216, Apr 2011. DOI: 10.3109/17453674.2011.566141. PMID: 21434784.
- ↑ S. Ferrari, P. Ruggieri, G. Cefalo, A. Tamburini i inni. Neoadjuvant chemotherapy with methotrexate, cisplatin, and doxorubicin with or without ifosfamide in nonmetastatic osteosarcoma of the extremity: an Italian sarcoma group trial ISG/OS-1. „J Clin Oncol”. 30 (17), s. 2112–2118, Jun 2012. DOI: 10.1200/JCO.2011.38.4420. PMID: 22564997.
- ↑ N. Marina i inni, MAPIE vs MAP as postoperative chemotherapy in patients with a poor response to preoperative chemotherapy for newly-diagnosed osteosarcoma: results from EURAMOS-1 (Paper 032), 2014 [dostęp 2016-04-29] [zarchiwizowane z adresu 2016-07-12].
- ↑ a b c Biermann i in. 2016 ↓, s. 21.
- ↑ a b c P.A. Meyers, C.L. Schwartz, M.D. Krailo, J.H. Healey i inni. Osteosarcoma: the addition of muramyl tripeptide to chemotherapy improves overall survival--a report from the Children’s Oncology Group. „J Clin Oncol”. 26 (4), s. 633–638, Feb 2008. DOI: 10.1200/JCO.2008.14.0095. PMID: 18235123.
- ↑ S. Hunsberger, B. Freidlin, M.A. Smith. Complexities in interpretation of osteosarcoma clinical trial results. „J Clin Oncol”. 26 (18), s. 3103-3104; author reply 3104-5, Jun 2008. DOI: 10.1200/JCO.2008.17.3484. PMID: 18565905.
- ↑ a b Rutkowski i Warzocha 2013 ↓, s. 451.
- ↑ S.S. Bielack, S. Smeland, J.S. Whelan, N. Marina i inni. Methotrexate, Doxorubicin, and Cisplatin (MAP) Plus Maintenance Pegylated Interferon Alfa-2b Versus MAP Alone in Patients With Resectable High-Grade Osteosarcoma and Good Histologic Response to Preoperative MAP: First Results of the EURAMOS-1 Good Response Randomized Controlled Trial. „J Clin Oncol”. 33 (20), s. 2279–2287, Jul 2015. DOI: 10.1200/JCO.2014.60.0734. PMID: 26033801.
- ↑ a b c Biermann i in. 2016 ↓, s. 54.
- ↑ B.A. Guadagnolo, G.K. Zagars, A.K. Raymond, R.S. Benjamin i inni. Osteosarcoma of the jaw/craniofacial region: outcomes after multimodality treatment. „Cancer”. 115 (14), s. 3262–3270, Jul 2009. DOI: 10.1002/cncr.24297. PMID: 19382187.
- ↑ a b Biermann i in. 2016 ↓, s. 52.
- ↑ D. Carrle, S.S. Bielack. Current strategies of chemotherapy in osteosarcoma. „Int Orthop”. 30 (6), s. 445–451, Dec 2006. DOI: 10.1007/s00264-006-0192-x. PMID: 16896870.
- ↑ a b V.H. Bramwell, M. Burgers, R. Sneath, R. Souhami i inni. A comparison of two short intensive adjuvant chemotherapy regimens in operable osteosarcoma of limbs in children and young adults: the first study of the European Osteosarcoma Intergroup. „J Clin Oncol”. 10 (10), s. 1579–1591, Oct 1992. PMID: 1403038.
- ↑ a b R.L. Souhami, A.W. Craft, J.W. Van der Eijken, M. Nooij i inni. Randomised trial of two regimens of chemotherapy in operable osteosarcoma: a study of the European Osteosarcoma Intergroup. „Lancet”. 350 (9082), s. 911–917, Sep 1997. DOI: 10.1016/S0140-6736(97)02307-6. PMID: 9314869.
- ↑ G. Bacci, A. Briccoli, S. Ferrari, A. Longhi i inni. Neoadjuvant chemotherapy for osteosarcoma of the extremity: long-term results of the Rizzoli’s 4th protocol. „Eur J Cancer”. 37 (16), s. 2030–2039, Nov 2001. PMID: 11597381.
- ↑ S. Ferrari, S. Smeland, M. Mercuri, F. Bertoni i inni. Neoadjuvant chemotherapy with high-dose Ifosfamide, high-dose methotrexate, cisplatin, and doxorubicin for patients with localized osteosarcoma of the extremity: a joint study by the Italian and Scandinavian Sarcoma Groups. „J Clin Oncol”. 23 (34), s. 8845–8852, Dec 2005. DOI: 10.1200/JCO.2004.00.5785. PMID: 16246977.
- ↑ I.J. Lewis, M.A. Nooij, J. Whelan, M.R. Sydes i inni. Improvement in histologic response but not survival in osteosarcoma patients treated with intensified chemotherapy: a randomized phase III trial of the European Osteosarcoma Intergroup. „J Natl Cancer Inst”. 99 (2), s. 112–128, Jan 2007. DOI: 10.1093/jnci/djk015. PMID: 17227995.
- ↑ K. Winkler, G. Beron, R. Kotz, M. Salzer-Kuntschik i inni. Neoadjuvant chemotherapy for osteogenic sarcoma: results of a Cooperative German/Austrian study. „J Clin Oncol”. 2 (6), s. 617–624, Jun 1984. PMID: 6202851.
- ↑ a b Perry 2008 ↓, s. 477.
- ↑ K. Winkler, G. Beron, G. Delling, U. Heise i inni. Neoadjuvant chemotherapy of osteosarcoma: results of a randomized cooperative trial (COSS-82) with salvage chemotherapy based on histological tumor response. „J Clin Oncol”. 6 (2), s. 329–337, Feb 1988. PMID: 2448428.
- ↑ a b Perry 2008 ↓, s. 478.
- ↑ P.A. Meyers, C.L. Schwartz, M. Krailo, E.S. Kleinerman i inni. Osteosarcoma: a randomized, prospective trial of the addition of ifosfamide and/or muramyl tripeptide to cisplatin, doxorubicin, and high-dose methotrexate. „J Clin Oncol”. 23 (9), s. 2004–2011, Mar 2005. DOI: 10.1200/JCO.2005.06.031. PMID: 15774791.
- ↑ M.M. Zalupski, C. Rankin, J.R. Ryan, D.R. Lucas i inni. Adjuvant therapy of osteosarcoma--A Phase II trial: Southwest Oncology Group study 9139. „Cancer”. 100 (4), s. 818–825, Feb 2004. DOI: 10.1002/cncr.20021. PMID: 14770440.
- ↑ P.A. Voûte, R.L. Souhami, M. Nooij, R. Somers i inni. A phase II study of cisplatin, ifosfamide and doxorubicin in operable primary, axial skeletal and metastatic osteosarcoma. European Osteosarcoma Intergroup (EOI). „Ann Oncol”. 10 (10), s. 1211–1218, Oct 1999. PMID: 10586339.
- ↑ a b c DeVita, Lawrence i Rosenberg 2008 ↓, s. 1822.
- ↑ a b Biermann i in. 2016 ↓, s. 55.
- ↑ Biermann i in. 2016 ↓, s. 56.
- ↑ J.C. Gentet, M. Brunat-Mentigny, M.C. Demaille, F. Pein i inni. Ifosfamide and etoposide in childhood osteosarcoma. A phase II study of the French Society of Paediatric Oncology. „Eur J Cancer”. 33 (2), s. 232–237, Feb 1997. PMID: 9135494.
- ↑ M. Berger, B. Massimo, G. Grignani, G. Giovanni i inni. Phase 2 trial of two courses of cyclophosphamide and etoposide for relapsed high-risk osteosarcoma patients. „Cancer”. 115 (13), s. 2980–2987, Jul 2009. DOI: 10.1002/cncr.24368. PMID: 19452540.
- ↑ O. Merimsky, I. Meller, G. Flusser, Y. Kollender i inni. Gemcitabine in soft tissue or bone sarcoma resistant to standard chemotherapy: a phase II study. „Cancer Chemother Pharmacol”. 45 (2), s. 177–181, 2000. DOI: 10.1007/s002800050027. PMID: 10663634.
- ↑ F. Navid, J.R. Willert, M.B. McCarville, W. Furman i inni. Combination of gemcitabine and docetaxel in the treatment of children and young adults with refractory bone sarcoma. „Cancer”. 113 (2), s. 419–425, Jul 2008. DOI: 10.1002/cncr.23586. PMID: 18484657.
- ↑ P. Van Winkle, A. Angiolillo, M. Krailo, Y.K. Cheung i inni. Ifosfamide, carboplatin, and etoposide (ICE) reinduction chemotherapy in a large cohort of children and adolescents with recurrent/refractory sarcoma: the Children’s Cancer Group (CCG) experience. „Pediatr Blood Cancer”. 44 (4), s. 338–347, Apr 2005. DOI: 10.1002/pbc.20227. PMID: 15503297.
- ↑ R.L. Saylors, K.C. Stine, J. Sullivan, J.L. Kepner i inni. Cyclophosphamide plus topotecan in children with recurrent or refractory solid tumors: a Pediatric Oncology Group phase II study. „J Clin Oncol”. 19 (15), s. 3463–3469, Aug 2001. PMID: 11481351.
- ↑ P.M. Anderson, G.A. Wiseman, A. Dispenzieri, C.A. Arndt i inni. High-dose samarium-153 ethylene diamine tetramethylene phosphonate: low toxicity of skeletal irradiation in patients with osteosarcoma and bone metastases. „J Clin Oncol”. 20 (1), s. 189–196, Jan 2002. PMID: 11773169.
- ↑ D.M. Loeb, E. Garrett-Mayer, R.F. Hobbs, A.R. Prideaux i inni. Dose-finding study of 153Sm-EDTMP in patients with poor-prognosis osteosarcoma. „Cancer”. 115 (11), s. 2514–2522, Jun 2009. DOI: 10.1002/cncr.24286. PMID: 19338063.
- ↑ P.M. Anderson, V. Subbiah, E. Rohren. Bone-seeking radiopharmaceuticals as targeted agents of osteosarcoma: samarium-153-EDTMP and radium-223. „Adv Exp Med Biol”. 804, s. 291–304, 2014. DOI: 10.1007/978-3-319-04843-7_16. PMID: 24924181.
- ↑ G. Grignani, E. Palmerini, P. Dileo, S.D. Asaftei i inni. A phase II trial of sorafenib in relapsed and unresectable high-grade osteosarcoma after failure of standard multimodal therapy: an Italian Sarcoma Group study. „Ann Oncol”. 23 (2), s. 508–516, Feb 2012. DOI: 10.1093/annonc/mdr151. PMID: 21527590.
- ↑ G. Grignani, E. Palmerini, V. Ferraresi, L. D’Ambrosio i inni. Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: a non-randomised phase 2 clinical trial. „Lancet Oncol”. 16 (1), s. 98–107, Jan 2015. DOI: 10.1016/S1470-2045(14)71136-2. PMID: 25498219.
- ↑ a b c d A.H. Aljubran, A. Griffin, M. Pintilie, M. Blackstein. Osteosarcoma in adolescents and adults: survival analysis with and without lung metastases. „Ann Oncol”. 20 (6), s. 1136–1141, Jun 2009. DOI: 10.1093/annonc/mdn731. PMID: 19153114.
- ↑ H.D. Dorfman, B. Czerniak. Bone cancers. „Cancer”. 75 (1 Suppl), s. 203–210, Jan 1995. PMID: 8000997.
- ↑ T.A. Damron, W.G. Ward, A. Stewart. Osteosarcoma, chondrosarcoma, and Ewing’s sarcoma: National Cancer Data Base Report. „Clin Orthop Relat Res”. 459, s. 40–47, Jun 2007. DOI: 10.1097/BLO.0b013e318059b8c9. PMID: 17414166.
- ↑ R.J. Grimer, S.R. Cannon, A.M. Taminiau, S. Bielack i inni. Osteosarcoma over the age of forty. „Eur J Cancer”. 39 (2), s. 157–163, Jan 2003. PMID: 12509946.
- ↑ A. Longhi, C. Errani, D. Gonzales-Arabio, C. Ferrari i inni. Osteosarcoma in patients older than 65 years. „J Clin Oncol”. 26 (33), s. 5368–5373, Nov 2008. DOI: 10.1200/JCO.2007.14.9104. PMID: 18809616.
- ↑ B. Carsi, M.G. Rock. Primary osteosarcoma in adults older than 40 years. „Clin Orthop Relat Res”, s. 53–61, Apr 2002. PMID: 11953595.
- ↑ M.L. Cowan, D.J. Monks, S.R. Raidal. Osteosarcoma in a woma python (Aspidites ramsayi). „Aust Vet J”. 89 (12), s. 520–523, Dec 2011. DOI: 10.1111/j.1751-0813.2011.00853.x. PMID: 22103954.
- ↑ K.E. Dittmer, A.F. French, D.J. Thompson, K.N. Buckle i inni. Primary bone tumors in birds: a review and description of two new cases. „Avian Dis”. 56 (2), s. 422–426, Jun 2012. DOI: 10.1637/9854-071911-Case.1. PMID: 22856206.
- ↑ B.K. Hartup, H. Steinberg. Osteosarcoma in an American robin (Turdus migratorius). „Avian Dis”. 40 (4). s. 938–940. PMID: 8980829.
- ↑ M. Fordham, K. Rosenthal, A. Durham, L. Duda i inni. Intraocular osteosarcoma in an Umbrella Cockatoo (Cacatua alba). „Vet Ophthalmol”. 13 Suppl, s. 103–108, Sep 2010. DOI: 10.1111/j.1463-5224.2010.00798.x. PMID: 20840098.
- ↑ S.M. Churgin, H. Steinberg, M. Ravi, B.K. Hartup. Sternal osteosarcoma in a blue crane (Anthropoides paradiseus). „J Zoo Wildl Med”. 44 (4), s. 1075–1078, Dec 2013. DOI: 10.1638/2012-0199R.1. PMID: 24450072.
- ↑ L.M. De Luca Bossa, G. Mennonna, L. Meomartino, O. Paciello i inni. Polyostotic Chondroblastic Osteosarcoma in a Kestrel (Falco tinnunculus). „J Avian Med Surg”. 29 (4), s. 336–339, Dec 2015. DOI: 10.1647/2014-052. PMID: 26771323.
- ↑ P. Mouser, A. Cole, T.L. Lin. Maxillary osteosarcoma in a prairie dog (Cynomys ludovicianus). „J Vet Diagn Invest”. 18 (3), s. 310–312, May 2006. PMID: 16789726.
- ↑ H. Tamaizumi, H. Kondo, H. Shibuya, M. Onuma i inni. Tail root osteosarcoma in a chipmunk (Tamias sibiricus). „Vet Pathol”. 44 (3), s. 392–394, May 2007. DOI: 10.1354/vp.44-3-392. PMID: 17491085.
- ↑ S. Simova-Curd, D. Nitzl, A. Pospischil, J.M. Hatt. Lumbar osteosarcoma in a chinchilla (Chinchilla laniger). „J Small Anim Pract”. 49 (9), s. 483–485, Sep 2008. DOI: 10.1111/j.1748-5827.2008.00584.x. PMID: 18631221.
- ↑ L.I. Dadone, D.P. Whiteside, S.R. Black, A. Remedios i inni. Nasal osteosarcoma and interstitial cell tumor in a Vancouver Island marmot (Marmota vancouverensis). „J Zoo Wildl Med”. 42 (2), s. 330–334, Jun 2011. DOI: 10.1638/2010-0159.1. PMID: 22946416.
- ↑ G.W. Salyards, U. Blas-Machado, S. Mishra, S.B. Harvey i inni. Spontaneous osteoblastic osteosarcoma in a Mongolian gerbil (Meriones unguiculatus). „Comp Med”. 63 (1), s. 62–66, Feb 2013. PMID: 23561939.
- ↑ J.G. Johnson, K. Kim, J. Serio, D. Paulsen i inni. Mandibular osteosarcoma in a nutria (Myocastor coypus). „J Zoo Wildl Med”. 45 (3), s. 723–726, Sep 2014. DOI: 10.1638/2014-0032R.1. PMID: 25314853.
- ↑ H. Kondo, M. Ishikawa, H. Maeda, M. Onuma i inni. Spontaneous osteosarcoma in a rabbit (Oryctolagus cuniculus). „Vet Pathol”. 44 (5), s. 691–694, Sep 2007. DOI: 10.1354/vp.44-5-691. PMID: 17846243.
- ↑ G. Mazzullo, M. Russo, P.P. Niutta, G. De Vico. Osteosarcoma with multiple metastases and subcutaneous involvement in a rabbit (Oryctolagus cuniculus). „Vet Clin Pathol”. 33 (2), s. 102–104, 2004. PMID: 15195269.
- ↑ J. Rodriguez-Ramos Fernandez, N.J. Thomas, R.R. Dubielzig, R. Drees. Osteosarcoma of the maxilla with concurrent osteoma in a southern sea otter (Enhydra lutris nereis). „J Comp Pathol”. 147 (2–3). s. 391–396. DOI: 10.1016/j.jcpa.2012.01.017. PMID: 22520807.
- ↑ H.L. Reid, S.L. Deem, S.B. Citino. Extraosseous osteosarcoma in a maned wolf (Chrysocyon brachyurus). „J Zoo Wildl Med”. 36 (3), s. 523–526, Sep 2005. DOI: 10.1638/04-043.1. PMID: 17312777.
- ↑ K.H. Plumlee, J.S. Haynes, K.W. Kersting, J.R. Thompson. Osteosarcoma in a cow. „J Am Vet Med Assoc”. 202 (1), s. 95–96, Jan 1993. PMID: 8420914.
- ↑ U. Braun, C.C. Schwarzwald, E. Forster, M. Becker-Birck i inni. Extraskeletal osteosarcoma of the thorax in a goat: case report. „BMC Vet Res”. 7, s. 55, 2011. DOI: 10.1186/1746-6148-7-55. PMID: 21929794.
- ↑ M.M. Williamson, A. Byrne. Mandibular osteosarcoma in a pig. „Aust Vet J”. 84 (6), s. 202–203, Jun 2006. PMID: 16821487.
- ↑ A.D. Tuttle, L. Frederico, K. Linder, C. Gunkel i inni. Pathological fracture of the ulna due to osteosarcoma in an Arabian camel (Camelus dromedarius). „Vet Rec”. 161 (1), s. 30–33, Jul 2007. PMID: 17617544.
- ↑ F. Jenner, M. Solano, J. Gliatto, S. Lavallee i inni. Osteosarcoma of the tarsus in a horse. „Equine Vet J”. 35 (2), s. 214–216, Mar 2003. PMID: 12638802.
- ↑ M.A. Livesey, I.W. Wilkie. Focal and multifocal osteosarcoma in two foals. „Equine Vet J”. 18 (5), s. 407–410, Sep 1986. PMID: 3464422.
- ↑ A.M. Nelson, D.C. Baker. Pedal osteosarcoma in a donkey. „Vet Pathol”. 35 (5), s. 407–409, Sep 1998. DOI: 10.1177/030098589803500510. PMID: 9754546.
- ↑ E. Momotani, H. Aoki, Y. Ishikawa, T. Yoshino. Osteosarcoma in the maxilla of a brown bear (Ursus arctos). „Vet Pathol”. 25 (6), s. 527–529, Nov 1988. PMID: 3212897.
- ↑ M. Liptovszky, E. Perge, V. Molnár, E. Sós. Osteoblastic osteosarcoma in a Grey Mouse Lemur (Microcebus murinus) – short communication. „Acta Vet Hung”. 59 (4), s. 433–437, Dec 2011. DOI: 10.1556/AVet.2011.030. PMID: 22079704.
- ↑ a b c M.J. Mezzles, E.J. Dick, M.A. Owston, C. Bauer. Osteosarcoma in Baboons (Papio spp). „Comp Med”. 65 (2), s. 144–149, Apr 2015. PMID: 25926401.
- ↑ a b Donald J. Meuten: Tumors in Domestic Animals. John Wiley & Sons, 2008, s. 264–265. ISBN 978-0-470-37670-6.
- ↑ a b c d e f g h i j Rafał Sapierzyński. Nowotwory narządu ruchu u psów i kotów. Część I. Kostniakomięsaki. „Życie Weterynaryjne”, 2005. [zarchiwizowane z adresu 2016-05-30].
- ↑ Joanna Morris, Jane Dobson: Small Animal Oncology. John Wiley & Sons, 2008, s. 80. ISBN 978-0-470-68013-1.
- ↑ Joanna Morris, Jane Dobson: Small Animal Oncology. John Wiley & Sons, 2008, s. 81. ISBN 978-0-470-68013-1.
- ↑ Joanna Morris, Jane Dobson: Small Animal Oncology. John Wiley & Sons, 2008, s. 87–88. ISBN 978-0-470-68013-1.
- ↑ Karen M. Tobias, Spencer A. Johnston: Veterinary Surgery: Small Animal: 2-Volume Set. Elsevier Health Sciences, 2013, s. 1162–1163. ISBN 978-0-323-26337-5.
- ↑ a b Joanna Morris, Jane Dobson: Small Animal Oncology. John Wiley & Sons, 2008, s. 87. ISBN 978-0-470-68013-1.
- ↑ a b Karen M. Tobias, Spencer A. Johnston: Veterinary Surgery: Small Animal: 2-Volume Set. Elsevier Health Sciences, 2013, s. 1173. ISBN 978-0-323-26337-5.
- ↑ Karen M. Tobias, Spencer A. Johnston: Veterinary Surgery: Small Animal: 2-Volume Set. Elsevier Health Sciences, 2013, s. 1173–1174. ISBN 978-0-323-26337-5.
Bibliografia
- Christopher D.M. Fletcher, K. Krishnan Unni, Fredrik Mertens: Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon: IARC Press, 2002.
- J. Sybil Biermann, Warren Chow, Douglas R. Adkins, Mark Agulnik i inni. Bone Cancer Version 2.2016. „NCCN”, 2016.
- P.G. Casali, J.Y. Blay, A. Bertuzzi, S. Bielack i inni. Bone sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. „Ann Oncol”. 25 Suppl 3, s. iii113-23, Sep 2014. DOI: 10.1093/annonc/mdu256. PMID: 25210081.
- Maciej Krzakowski, Piotr Potemski, Krzysztof Warzocha, Pior Wysocki: Onkologia kliniczna. T. II. Via Medica, 2015. ISBN 978-83-7599-796-5.
- Mięsaki kości. W: Piotr Rutkowski, Krzysztof Warzocha: Zalecenia postępowania diagnostyczno-terapeutycznego w nowotworach złośliwych 2013 rok. VM Media Sp z o.o., 2013. ISBN 978-83-7599-594-7.
- Vincent T. DeVita, Theodore S. Lawrence, Steven A. Rosenberg: Devita, Hellman & Rosenberg’s Cancer: Principles & Practice of Oncology. Wyd. 8. Lippincott Williams & Wilkins, 2008. ISBN 978-0-7817-7207-5.
- Christopher D.M. Fletcher: Diagnostic Histopathology of Tumors. Elsevier Health Sciences, 2007. ISBN 978-0-7020-3205-9.
- Marilyn M. Bui, Leslie G. Dodd: Atlas of Soft Tissue and Bone Pathology: With Histologic, Cytologic, and Radiologic Correlations. Demos Medical Publishing, 2014. ISBN 978-1-61705-199-9.
- K. Krishnan Unni, Carrie Y. Inwards: Dahlin’s Bone Tumors: General Aspects and Data on 10,165 Cases. Lippincott Williams & Wilkins, 2010. ISBN 978-0-7817-6242-7.
- Giulia Ottaviani, Norman Jaffe: The etiology of osteosarcoma. W: Norman Jaffe, Oyvind S. Bruland, Stefan Bielack: Pediatric and Adolescent Osteosarcoma. Springer Science & Business Media, 2010. ISBN 978-1-4419-0284-9.
- Michael J. Perry: The Chemotherapy source book. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins, 2008. ISBN 978-1451101454.
- Bogdan Czerniak: Dorfman and Czerniak’s Bone Tumors. Elsevier Health Sciences, 2015. ISBN 978-0-323-09159-6.
![]() Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.
Przeczytaj ostrzeżenie dotyczące informacji medycznych i pokrewnych zamieszczonych w Wikipedii.
Media użyte na tej stronie

The Star of Life, medical symbol used on some ambulances.
Star of Life was designed/created by a National Highway Traffic Safety Administration (US Gov) employee and is thus in the public domain.Autor: Nephron, Licencja: CC BY-SA 3.0
Micrograph of an osteosarcoma, a malignant primary bone tumour. H&E stain.
Osteosarcomas, by definition, form osteoid (the organic extracellular component of bone) which is pink, bland/homogenous on H&E staining.
Features of osteosarcoma seen in image:
- High density of spindle cells with malignant features (e.g. nuclear membrane irregularies, marked nuclear size differences, mitoses) surrounded by delicate strands of osteoid.
- Large (multinucleated) osteoclast-like giant cells (seen occasionally in osteosarcomas).
- Title Osteosarcoma (Bone Cancer)
- Description Human malignant osteosarcoma (bone cancer) cells taken by aspirant from a leg mass. Pap stain and magnified to 400x.
- Topics/Categories Cancer Types -- Bone Cancer Cells or Tissue -- Abnormal Cells or Tissue
- Type Color, Photo
- Source Dr. Lance Liotta Laboratory
Autor: Nephron, Licencja: CC BY-SA 3.0
High magnification micrograph of small cell osteosarcoma. H&E stain.
Small cell osteosarcoma, like other osteosarcomas has osteoid, the extracellular organic component of bone. On H&E stained sections, osteoid has a pink, homogeneous appearance and surrounds osteoblasts or osteosarcoma cells.
Related images

Intermed. mag.

High mag.

Very high mag.



Another case of osteosarcoma

Low mag.
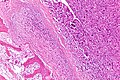
Intermed. mag.

High mag.
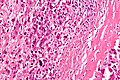
Very high mag.




Autor: unknown, Licencja: CC BY 4.0
Osteosarcoma of the thigh bone, Egyptian, 5th Dynasty
General Collections
Keywords: palaeopathology; egyptology; oncology; G. Elliot Smith,; Warren R. Dawson,
Autor: User:Ajimsha619, Licencja: CC BY-SA 3.0
codman triangle
Autor: Nephron, Licencja: CC BY-SA 3.0
High magnification micrograph of a high-grade osteosarcoma. H&E stain.
By definition, osteosarcomas form osteoid (the organic extracellular component of bone), which is pink, bland/homogenous on H&E staining.
Related images
Low mag.
Intermed. mag.
High mag.
Very high mag.
Autor: Nephron, Licencja: CC BY-SA 3.0
Very high magnification micrograph of a high-grade osteosarcoma. H&E stain.
By definition, osteosarcomas form osteoid (the organic extracellular component of bone), which is pink, bland/homogenous on H&E staining.
Related images
Low mag.
Intermed. mag.
High mag.
Very high mag.
Autor: unknown, Licencja: CC BY 4.0
Black and white photograph of a male patient, aged 49 years, with a very large osteo-sarcoma of the right humerus. The tumour had existed for eight years, but had increased very rapidly during the last year. It was removed by amputation at the shoulder joint, and the patient made a good recovery. The limb, with tumour, weighed 33lbs, after removal. Photograph taken by John Howell Griffiths whilst a student at St Bartholomew's Hospital Medical School.
Medical Photographic Library
Keywords: St Bartholomew's Hospital Photographic Society